
Jesús Alberto Segovia-Cruz a, Valeria Souza b, Yuridia Mercado-Flores a, Miguel Ángel Anducho-Reyes a, Genaro Vargas-Hernández c, Alejandro Téllez-Jurado a, *
a Universidad Politécnica de Pachuca, Laboratorio de Agrobiotecnología, Carretera Pachuca-Ciudad Sahagún Km. 20, Ex-Hacienda de Santa Bárbara, 43830 Zempoala, Hidalgo, Mexico
b Universidad Nacional Autónoma de México, Instituto de Ecología, Departamento de Ecología Evolutiva, Tercer Circuito s/n, Ciudad Universitaria, Coyoacán, 04510 Ciudad de México, Mexico
c Universidad Politécnica de Pachuca, Laboratorio de Tecnología de Compuestos Bioactivos, Carretera Pachuca-Ciudad Sahagún Km. 20, Ex-Hacienda de Santa Bárbara, 43830 Zempoala, Hidalgo, Mexico
*Corresponding author: alito@upp.edu.mx (A. Téllez-Jurado)
Received: 16 September 2022; accepted: 9 April 2023
Abstract
This research studied the thermophilic microbial composition of samples from 4 areas of the Tecozautla geyser, Hidalgo, Mexico. The samples were: sediments (GD), salt deposits (GA), and microbial mats (GB and GC). The 16S rRNA gene amplicons were sequenced, obtaining 1,425,506 readings. Bioinformatic analysis identified 32 phyla in the 4 samples being the most representative for the GA: Armatimonadetes, Chloroflexi, Cyanobacteria, and Thermi. For the GB, they were: Proteobacteria, Bacteroidetes, Cyanobacteria, Spirochaetes, Thermi, and Firmicutes. For the GC the observed phyla were: Chloroflexi, Armatimonadetes, Proteobacteria, Cyanobacteria, and Acidobacteria. Finally, for the GD, the most abundant phyla were Chloroflexi, Cyanobacteria, Armatimonadetes, Proteobacteria, and Nitrospirae. The metabolic functionality of the microbial communities present in the samples was inferred using the 16S rRNA amplicons.
Keywords: 16S rRNA; Thermophilic microbial diversity; Microbial mats; Metabolic functionality
© 2023 Universidad Nacional Autónoma de México, Instituto de Biología. This is an open access article under the CC BY-NC-ND license
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Diversidad microbiana del géiser de Tecozautla, México
Resumen
En esta investigación, se estudió la composición termofílica microbiana de 4 áreas del géiser de Tecozautla, Hidalgo, México. Las muestras se tomaron de los sedimentos (GD), depósitos de sales (GA) y de tapetes microbianos (GB y GC). Para la identificación, se utilizó la secuenciación 16S rARN obteniéndose 1,425,509 lecturas. El análisis bioinformático de las 4 muestras permitió la identificación de 32 phyla siendo los más representativos: Armatimonadetes, Chloroflexi, Cyanobacteria y Thermi para la muestra GA. Para la muestra GB fueron: Proteobacteria, Bacteroidetes, Cyanobacteria, Spirochaetes y Firmicutes. Para la muestra GC se detectaron los phyla: Chloroflexi, Armatimonadetes, Proteobacteria, Cyanobacteria y Acidobacteria. Finalmente, para la muestra GD, más abundantes fueron: Chloroflexi, Cyanobacteria, Armatimonadetes, Proteobacteria y Nitrospirae. Se infirió a través de los amplicones 16S rARN la funcionalidad metabólica de las comunidades microbianas.
Palabras clave: 16S rRNA; Diversidad micriobiana termofílica; Tapetes microbianos; Funcionalidad metabólica
© 2023 Universidad Nacional Autónoma de México, Instituto de Biología. Este es un artículo Open Access bajo la licencia CC BY-NC-ND
Introduction
The term “extremophile” applies to organisms capable of actively growing in difficult or hard conditions that would be lethal to most organisms on the planet (Atanasova et al., 2021). Most of the microorganisms capable of growing in extreme conditions of temperature, pH, pressure, and radiation meet the characteristics of being known as extremophilic microorganisms (Canganella & Wiegel, 2011). The evolutionary adaptations that allow them to survive such extreme conditions include modifications in their cellular components, such as the increase in their electrostatic interactions, hydrogen bonds, and hydrophobic interactions in their proteins, to mention a few (Reed et al., 2013). Several types of extremophilic microorganisms can be classified depending on the conditions of the environment where they grow: high-temperature, high salinity, few nutrients, and alkaline and acidic pH, mainly. For example, thermophilic microorganisms grow above 45 °C, and hyperthermophiles grow above 80 °C (Canganella & Wiegel, 2011). Thermophilic organisms can be distributed in different parts of the planet, mainly in places with volcanic activity, such as hot springs, underwater hydrothermal vents, abyssal thermal openings, and geysers.
Several studies have been carried out to determine the microbial communities of hot springs in different parts of the world. Uribe-Lorio et al. (2019) observed that Cyanobacteria and Chloroflexi are the predominant microbial communities in hot springs located in Costa Rica with a temperature of 37 °C and pH of 7.5. The bacterial composition changes as the temperature increases and the pH of the water decreases, finding that the most common phyla are Choloflexi and Deinococcus. For their part, Mackenzie et al. (2013) observed that the largest bacterial groups present in hot springs in Patagonia were Cyanobacteria and Bacteroidetes and the order Thermals. Yasir et al. (2020), in a study carried out in the hot springs of Al Aridhah (Saudi Arabia), determined that the phylum Chloroflexi was the most abundant in waters with a temperature of 48 °C. Prieto-Barajas et al. (2018) studied the microbial diversity of hot springs in the Araró region (Mexico), finding that the main phyla were: Proteobacteria, Chloroflexi, and Cyanobacteria. In hot springs with alkaline pH, Proteobacteria and the Bacteroidetes were dominant (Stom et al., 2022). It has also been found that the chemical composition of the hot springs defines the composition of the microbial mats, as in the case of the Jinata Onsen hot springs located on the island of Shikinejima, Japan. These hot springs are composed of marine waters with various redox potential gradients and high concentrations of iron ions that favor the presence of oxygenic bacteria such as the Cyanobacteria, and their presence decreases when iron toxicity decreases, favoring other bacterial phyla such as Chloroflexi and Calditrichaeota (Ward et al., 2019).
Most microorganisms that grow in extreme environments can associate and, on many occasions, secrete exopolysaccharides whose function is to allow adherence to solid surfaces. These associations generate complex ecological groups (Nicolaus et al., 2010). These mats, in turn, fulfill the function of protecting microorganisms through a physical barrier, contributing to water retention and, due to their anionic nature, allowing the exchange of metal cations and nutrients (Costa et al., 2018).
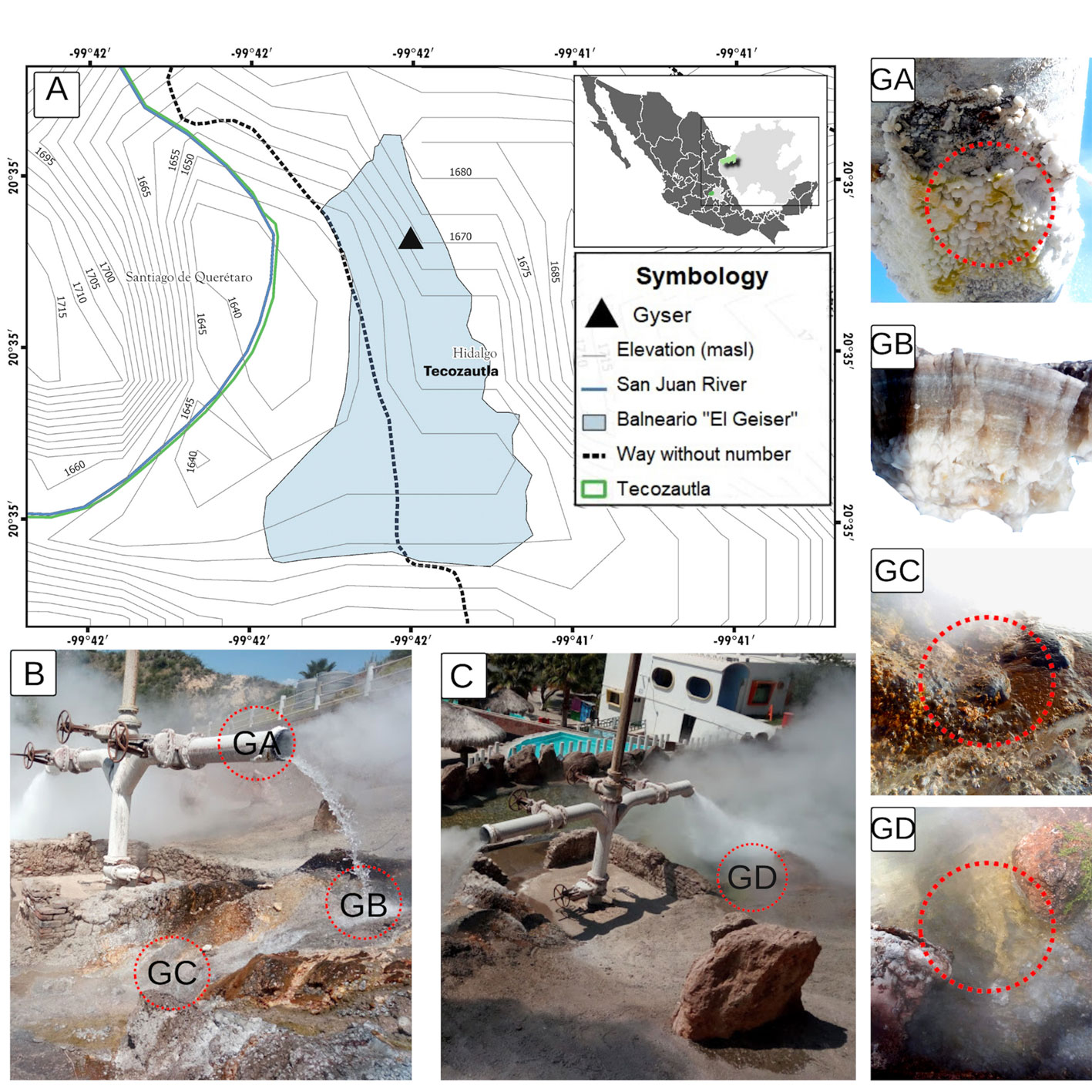
The word “geyser” comes from the old Icelandic verb, “gjose”, which means to erupt. It specifically refers to a hot water tank that intermittently and explosively expels part or all its contents (Rinehart, 1980), as is the case with the Tecozautla geyser, located in the municipality of the same name in the state of Hidalgo, Mexico. The geyser belongs to the Pathé geothermal zone, located on the border between Hidalgo and Querétaro, about 35 km in a straight line to the northwest of the municipality of San Juan del Río, Querétaro, and 20 km north of the town of Huichapan, Hidalgo. The area belongs to the physiographic province of the Mexican Volcanic Belt, very close to its limits with the Sierra Madre Oriental (Hiriart et al., 2011). This work aimed to study the microbial communities in the microbial mat formed from the hot springs from Tecozautla geyser.
Materials and methods
The Tecozautla geyser (TG) is at an altitude of 1,665 m (20°34’40.8” N, 99°41’35.0” W) within the spa known as “El Geiser” in the community of Uxdejhé, belonging to the municipality of Tecozautla, Hidalgo, Mexico. Samples were taken from 4 zones within the geyser: salt deposits (GA), microbial mats I (GB), microbial mats II (GC), and sediments + microbial mat (GD), recording temperatures and pH of the sampling zones. The collected samples were placed in sterile 50 mL Falcon tubes and transported in containers with ice. The samples were stored at -20 °C until analysis.
Twenty-three physicochemical parameters of the geyser water were determined: conductivity, pH, TSD, Cl–, NO3-, NO2-, HCO3-, SO42-, ammonia N, Al, As, Ba, Cd, CN-, Cu, Cr, Fe, Mn, Pb, Zn, active substances to methylene blue (SAAM), Na+ and Hg, according to the NOM-127-SSA1-1994. The samples were analyzed in the Academic Area of Chemistry of the Autonomous University of the State of Hidalgo, Mexico. The total sulfur was determined by emission spectroscopy with inductive plasma coupling (ICP) in a Perkin Elmer model Optima 8300 XL equipment. All the water samples to be analyzed were taken to 25 mL with deionized water (18 µS/cm). To determine the total sulfur, a standard sulfur stock solution was prepared at a final elemental sulfur concentration of 50 mg/L in 5% HNO3. 2 to 10 mg/L dilutions were made and read in a spectrophotometer (Thermo) at 182 nm for the calibration curve.
Two DNA extraction methodologies were used since the samples presented different compositions, such as high exopolysaccharides and salts, making them difficult to obtain. The extraction of metagenomic DNA from the GA, GB, and GC samples was carried out with the following methodology. The lysis of the samples was carried out by adding 0.5 g of glass beads (425-600 μm), 0.5 mL of DNA, 600 μL of a sodium phosphate solution (120 mM, pH 8), 1% (p/v) of PVP (polyvinylpyrrolidone), 400 µL of Tris-phenol solution (pH 8), 40 µL of SDS (20%); followed by 3 cycles of shake-ice on a Tissue Lyses LT (QIAGEN) for 30 seconds at maximum speed (50 Hz). The samples were then centrifuged at 13,000 rpm for 1 min. The supernatant was transferred to an HTP (hydroxyapatite) column by centrifuging at 1,500 rpm for 2 min, followed by 3 continuous washes of the column with 500 µL of 120 mM sodium phosphate pH 7.2, each time centrifuging at 1,500 rpm for 6 min. The DNA was eluted by adding 400 µL of a dipotassium phosphate solution (300 mM, pH 7.2) and centrifuging at 1,500 rpm for 6 min. Once eluted, the DNA solution was transferred to a G50 Sephadex column and centrifuged at 13,000 rpm for 1 min. Subsequently, the DNA was precipitated overnight at -20 °C with the addition of 40 μL of a sodium acetate solution (3 M) and 1 mL of absolute cold ethanol. The DNA was recovered by centrifugation at 13,000 rpm for 10 min. Subsequently, the pellet was washed with 70% alcohol and resuspended in 20-50 μL of DNAase-free water.
For the extraction of metagenomic DNA from the GD sample. To obtain a homogeneous mixture, approximately 0.5 g of the sample were macerated with liquid nitrogen and extraction buffer (100 mM Tris-HCl, 20 mM NaCl, 100 mM EDTA pH 8, 1% (p/v) PVP), with 1% cetyltrimethylammonium bromide (CTAB) at 6%, following 3 continuous freeze-thaw cycles with liquid nitrogen and 65 °C. Subsequently, the samples were incubated for 30 min at 37 °C with lysozyme (30 mg/mL). Then 0.2 mL of 10% sodium dodecyl sulfate (SDS) and proteinase K (10 mg/mL) were added for 2 h at 65 °C. The nucleic acids were extracted twice with a solution of phenol: chloroform: isoamyl alcohol (25:24:1) and once with chloroform: isoamyl alcohol (24:1), recovering the supernatant liquid after each centrifugation (10,000 x g for 10 min). The DNA was precipitated with 0.6 volumes of cold isopropanol, 3 M sodium acetate (1/10 v), and centrifuged at 22,700 x g for 15 min at 4 °C. The DNA pellet obtained was washed with 7% cold ethanol, centrifuged, and the excess alcohol was allowed to evaporate. The DNA was resuspended in 30 μL of DNAase-free water. Finally, the commercial kit Zymo BIOMICS DNA (Zymo) was used to purify the DNA. The DNA quality was verified in a 0.8% agarose gel electrophoresis and stained with EpiQuick DNA Stain (10 μL/100 mL). DNA concentrations were determined using a Nanodrop Lite Spectrophotometer (Thermo Scientific).
16S rRNA sequencing. The purified DNA samples were used as a template to amplify the 16S rRNA gene using primers from the V3-V5 regions, according to the Illumina MiSeq protocol. The first universal 357-F (5´-CTCCTACGGGAGGCAGCAG -3´) and CD-R (5´ CTTGTGCGGGCCCCCGTCAATTC-3´) (Rudi et al., 1997) were used.
For the GA, GB, and GC samples a 2×150 run format was used, and for the GD sample 2×300. To carry out the amplification, the reaction mixture was adjusted to a final reaction volume of 25 μL with 5 μL (10 μM) of primer F, 5 μL (10 μM) of primer R, 2.5 μL of DNA (5 ng/μL in 10 mM Tris pH 8.5), 12.5 μL of 2xKAPA HiFi HotStart Ready Mix. The PCR consisted of an initial denaturation at 95 °C (3 min), followed by 25 cycles for denaturation (95 °C, 30 s) alignment (55 °C, 30 s), and extension (72 °C, 30 s). A final extension to 72 °C, 5 min was used (https://www.illumina.com/content/dam/illumina- support/documents/documentation/chemistry_documentation/16s/ 6s-metagenomic-library-prep-guide-15044223-b.pdf). The Illumina libraries were purified with the DNA Clean & Concentrator purification kit (Zymo Research, Irvine, CA, USA). The DNA sequencing was done in the National Laboratory of Genomics for Diversity (LANGENBIO, CINVESTAV-Irapuato, México).
Analysis of amplicon readings. TagCleaner was used for the processing of the reads obtained to remove primers and ambiguous bases, consecutively Trimmomatic was used to filter the sequences, conserving those that had a length greater than 100 bp ≤ Q20 in the forward direction of the V3 region, due to their quality and abundance, in addition to the fact that they could not be paired because they were very distant from each other.
QIIME2 v2019.1 software was used, and the sequences were imported with Casava 1.8 single-end demultiplexed fastq, using a mapping file (.tsv) with the information of the samples. The analysis started with the DADA2 option, which consists of the purification and trimming of the sequences, carried out at 100 bp to preserve the greatest amount of information and eliminate errors in the sequencing. Identifying and eliminating the chimeric sequences and the “borderline chimeras” were carried out using the uchime-de novo method, grouping them de novo in operative taxonomic units (OTU’s) with a 97% similarity threshold, using the VSEARCH algorithm for both cases. The taxonomic assignment of the sequences was carried out using a Bayesian classifier with the complement q2-feature-classifier using the Greengenes database (gg-13-8-99-nb-classifier.qza) (Almeida et al., 2018). Additionally, graphs were made to describe the relative abundance at different taxonomic levels using ggplot2 in the R software (https://www.r-graph-gallery.com).
The diversity analyses were carried out by taking the sampling depth (rarefaction) as a parameter, starting from the sample with the lowest readings obtained. The alpha diversity and the observed OTU’s metrics, Chao 1, Shannon, and Simpson, were determined. In addition, Good coverage was determined (www.qiime2.org). Beta diversity was determined with qiime and plotted with Emperor. Principal coordinate analysis (PCoA) and Bray-Curtis method for decreased distance matrices were utilized. The MetaCoMET web tool was used to determine the central microbiome using an OTU table in BIOM v2.1.0 format containing the community abundance data and its taxonomy (https://probes.pw.usda.gov/MetaCoMET).
Prediction of metabolic functions was performed using PICRUSt. The analysis began with the single-end.qza file from where the chimeric sequences and the “borderline chimeras” were eliminated, using the uchime-de novo method, the sequences were grouped to closed references in operative taxonomic units (OTU’s) at 97% identity. The generated file was exported to the BIOM v2.1.0 format, and the taxonomic assignment to 97% identity with Greengenes data was attached. It was then uploaded to the PICRUSt platform, (http://galaxy.morganlangille.com/), where the functional metabolic prediction was performed (https://forum.qiime2.org/t/how-to-create-a-feature-table-with-qiime2-for-picrust-with-the-taxonomic-assignment/2526/6).
Results
The location of the TG is shown in figure 1A. The collection was carried out in June. The sampling areas are shown in figures 1B, C. The samples were collected in 4 areas of the TG in 2021. The GA sample was located on the tube of the outlet water of the geyser; it had a composition with a high content of white salts with green areas, registering a temperature of 94 °C (Fig. 1B: GA). The GB sample corresponds to a microbial mat found in the soil directly receiving the water from the geyser; it had white, brown, and green colors and presented a rigid and fibrous structure (65 °C) (Fig. 1B: GB). The GC sample corresponded to a microbial mat of reddish, yellow, green, pink, white, and brown color, with a semisolid structure (muddy) with small fibrous sections, found in a heat flux of 61.5 °C on a rock (Fig. 1B: GC). Finally, the GD sample corresponded to the sediments and microbial mats submerged in the left outlet of the geyser, with constant turbulence due to the continuous outlet of water (62 °C) (Fig. 1C: GD).
The results obtained from the chemical composition of the TG are presented in Table 1. It was observed that the water presented a conductivity of 1,215 μmoh/cm and 82 mg/L of total dissolved solids. The pH ranged between 9.24 and 10, indicating alkaline waters. Mainly sulfur (47 ± 2.51 mg/L), sodium (195 mg/L), and chlorides (156.33 mg/L) were detected. Elements such as Al, As, Ba, Cd, Cr, Mn, Pb, Zn, and Hg were not detected. The water temperature at the sampling time was 94 °C at the source. Finally, sulfates (28.6 mg/L), carbonates (16.29 mg/L), and a minimum concentration of nitrates (0.2 mg/L) were detected.
Taxonomic assignments of the readings and thermophilic diversity. The sequencing of the 4 samples yielded 5,660,646 raw paired-end (PE) readings, of which 2,830,823 were found in the forward direction; these sequences were carried out with the bioinformatic analysis. After removal of the primers with TagCleaner and quality filtering with Trimmomatic, 1,589,850 readings were obtained. DADA2 was used for removing chimeras, borderline chimeras, and clustering of sequences de novo at 97% similarity: obtaining a total of 1,425,506 readings, of which 1,178,144 corresponded to GA, 1,668 to GB, 43,769 to GC, and 201,925 to GD. The OTUs obtained for each sample were 434, 75, 130, and 648 for GA, GB, GC, and GD, respectively.
In the 4 samples, 30 bacterial and 2 archaeal phyla (in GA and GD), 70 classes, 79 orders, 72 families, and 44 genera were identified, and 2% for sequences not classified in GA and 0.10% for GD was observed. In addition, bacteria without taxonomic assignment were found at 26.75%, 8.40%, 5.99%, and 12.83%, for the GA, GB, GC, and GD samples, respectively. Figure 2 shows the relative abundances at the phylum level and class of the 4 areas sampled (Fig. 2A, B).
Thermophilic diversity in the TG. The analysis of the relative abundance of thermophilic microorganisms from the TG indicated high percentages of SJA-176 class detected in the GC and GD samples. In contrast, class Fimbriimonadia was found in the GC and GD.
The phylum Cyanobacteria is present in all samples of the TG, with relative abundances of 3.27%, 20.24%, 7.01%, and 17.13% for GA, GB, GC, and GD, respectively (Fig. 2A). The class Gloeobacterophycideae is present only in the GA sample, while Nostocophycideae in the GC sample and Oscillatoriophycideae in the GA, GB, and GC samples. Finally, the Synechococcophycideae are present in all samples (Fig. 2B). The organisms found in the TG at the genus level within the phylum Cyanobacteria were: Gloeobacter (GA), Leptolyngbya (GA and GB), and Pseudanabaena (GA, GC, and GD) (Fig. 2C).
Another abundant group in the TG samples was the phylum Chloroflexi. The class Thermomicrobia only was found in GD. Unidentified microorganisms were found at the class level with 0.62%, 0.14%, and 0.06% for GA, GC, and GD, respectively. The phylum Thermus was identified in GA, GB, GC, and GD. The Proteobacteria were among the most abundant members, with a presence in all the samples. In the TG all classes of Proteobacteria were identified: Alpha, Beta, Delta, Epsilon, and Gamma, which shows the great diversity in the microenvironments analyzed. The phylum Bacteroidetes was found in all the samples analyzed from the TG.
Other taxonomic groups identified
The Acidobacteria, Actinobacteria, Aquificae, Chlorobi, Dictyoglomi, Firmicutes, Gemmatimonadetes, Nitrospirae, Planctomycetes, Spirochaetes, Synergistetes, Tenericutes, Thermotogae, and Verrucomicrobia phyla were identified in the TG. Microorganisms with unknown characteristics were also found, such as TM7, WS6, OC31, OD1, OP1, OP11, OP9, OctSpA1-106, NKB19, and GAL15, with relative abundances that do not exceed 1%. The archaeal phyla found in the TG were Crenarchaeota and Euryarchaeota in the GA and GD samples.
Lower abundance percentages were observed for the relative abundances at the genus level because some sequences could not be classified at this level. Figure 2C shows the relative abundances of the microorganisms with the highest incidence.
In the GA sample, the main genera found were: Caldilinea, Thermus, Gloeobacter, and Truepera. The most abundant genera in the GB sample were Porifericola, Thermus, Halanaerobium, Blvii28, and Leptonema. The GC sample presented high abundances for: Roseiflexus, Candidatus Chloracidobacterium, Thermodesulfovibrio, and Spirochaeta. For the GD sample, some of the most abundant genera were: Roseiflexus, Chloroflexus, Thermodesulfovibrio, and Thermus. The genera found in the 4 samples belong to Hydrocarboniphaga (GA), Rhodovulum (GC), Novosphingobium, Acinetobacter, Geothermobacterium, and Desulfomicrobium (GD).
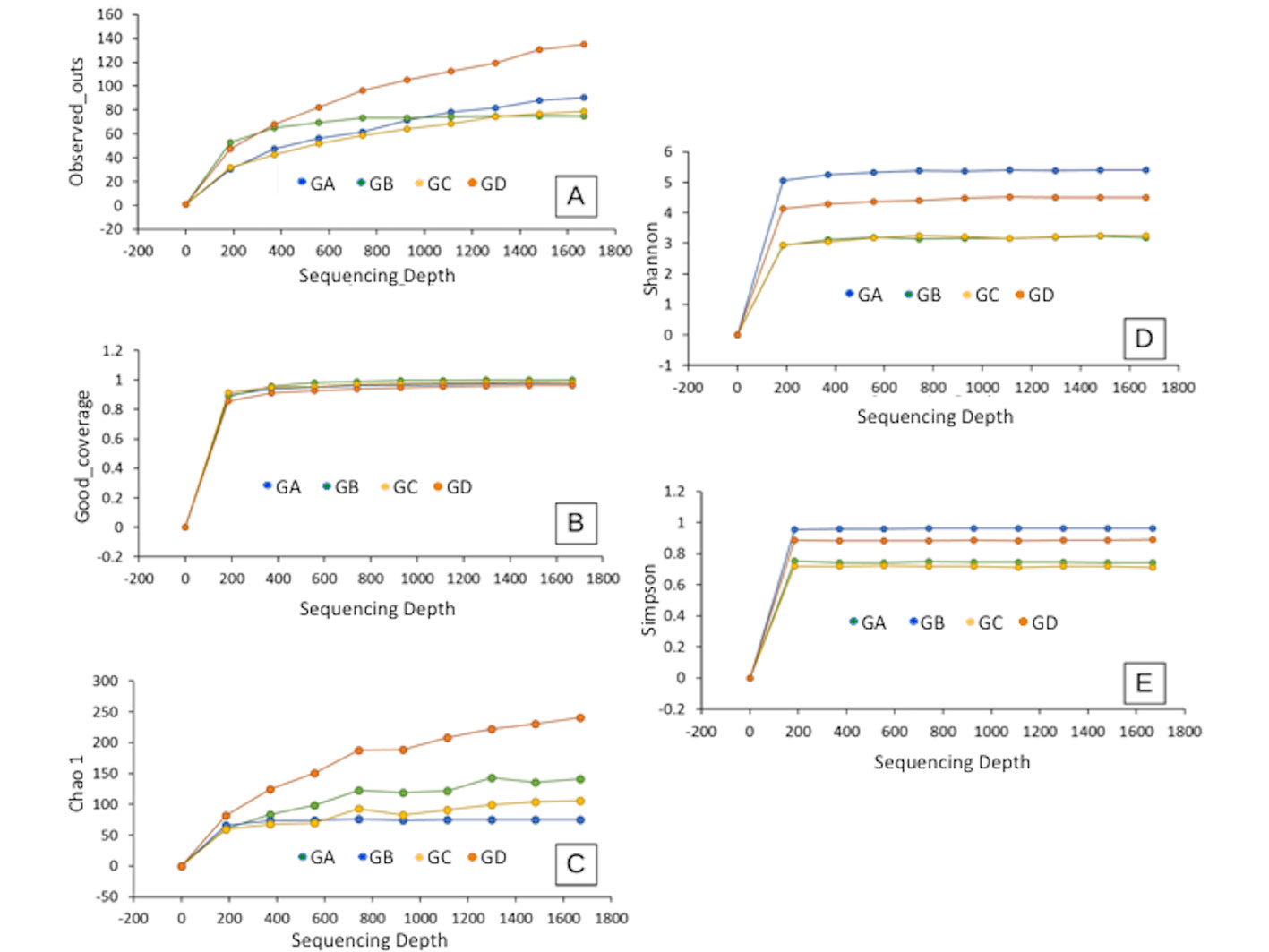
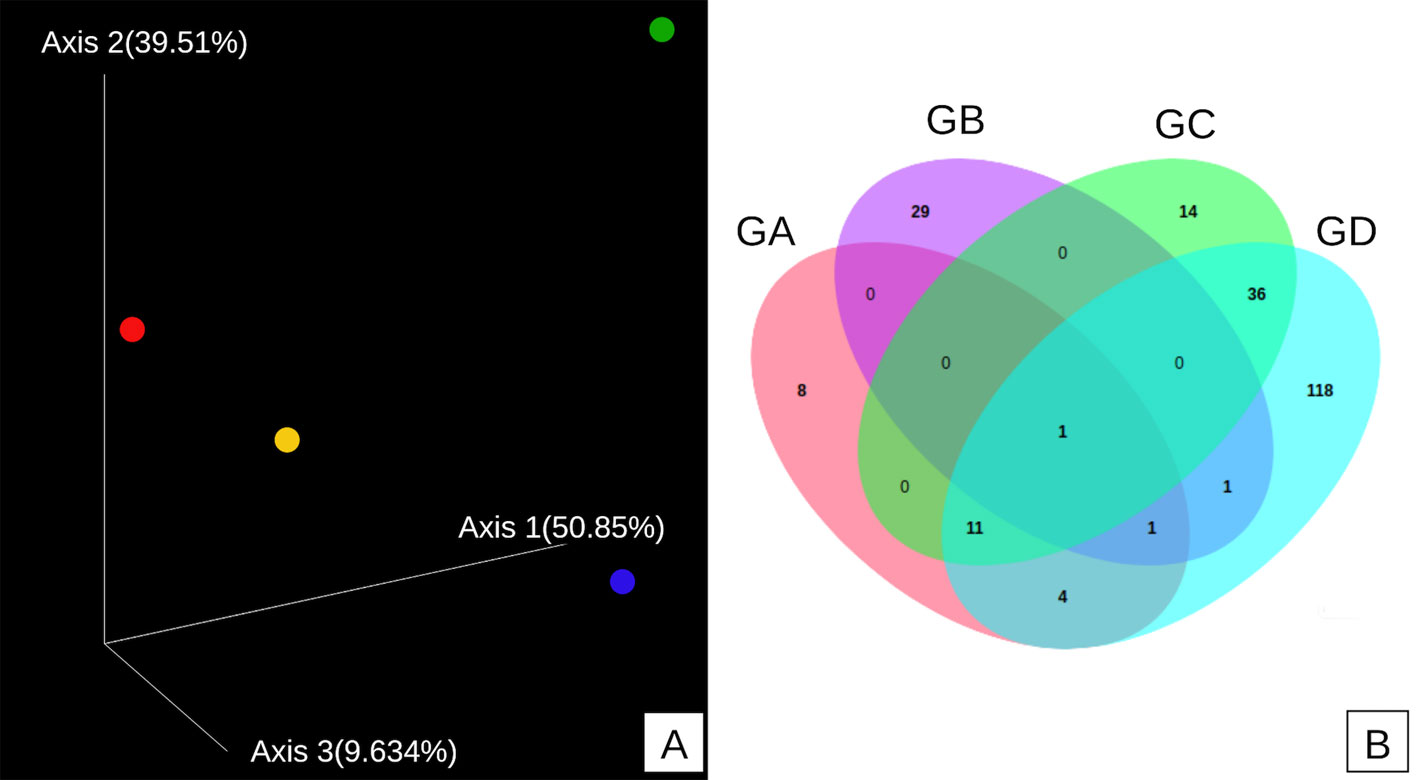
The diversity and richness of the samples were analyzed through the Chao1, Shannon, Simpson, OTUs, and Good coverage indices. Alpha diversity was higher in the GD sample (Fig. 3A), and sampling was acceptable in all samples according to Good coverage (Fig. 3B). The highest richness of microorganisms was observed in the GD sample (Chao1 index) (Fig. 3C). In contrast, the highest species diversity was observed in the GA sample (Shannon-Weiner index) (Fig. 3D), which agrees with what was observed with the Simpson index, where the GA sample was the most diverse one. In Figure 4A it is observed that the 4 samples are separated from each other; that is, none of the samples are superimposed. The Venn diagram of the central microbiome (Fig. 4B) shows the set of OTUs shared by the 4 samples collected from the TG. It is observed that all the samples share 1 OTU, which is related to unclassified bacterial diversity. The GA, GC, and GD samples share 11 OTUs, and the GA, GB, and GD samples share 1 OTU. The GA and GD samples share 4 OTUs, GB and GD share 1 OTU, GC, and GD share abundant diversity among themselves with 36 OTUs, possibly due to their physical proximity (sampling location). Each sample presents many OTUs they do not share; GA does not share 8 OTUs, GB 29 OTUs, GC 14 OTUs, and GD 118 OTUs. This result agrees with what was observed in beta diversity; that is, even though the physical location of each sample is relatively close, there are large differences in microbial diversity.
The functional profiles of the microbiome were predicted based on the 16S rRNA gene using the OTU table of assigned taxa and their relative distribution. On the other hand, using existing genomes and PRICUSt and KEGG analysis, it was possible to identify the main functional metabolic pathways present in microbial mats that allow the survival of microbial communities. The results obtained from the 4 samples of the TG presented similar functions to each other for level 2 (Fig. 5A). The highest abundances were the pathways related to membrane transport, carbohydrate metabolism, amino acid metabolism, energy metabolism, replication and repair, cofactor, and vitamin metabolism. These results suggest the main metabolic pathways of microorganisms in the TG, but at a not-so-specific level.

The most abundant metabolic pathways obtained for level 3 shown in Figure 5B, were like each other, predominating ABC membrane transport, DNA repair, protein recombination, prediction of general functions, membrane transport, metabolisms of the methane, and purine metabolism. Oxid phosphorylation stands out from energy metabolism, followed by routes intervening in carbon fixation and methanogenic metabolism (Fig. 5C).
Discussion
The chemical composition of hot springs is highly variable. It depends on the depths and the different paths they take to the surface (Nimick et al., 1998), implying that the concentrations of minerals and their presence change over time. The chemical composition of the hot spring waters of the TG differ from that described by Núñez-Benitez (2007), who reported the presence of Arsenic (43.77 mg/L) and silica (112.65 mg/L) in these same waters, while these salts were not detected in our study. Tamazawa et al. (2012) indicated that ions (such as SO4-2) could affect the morphology of microorganisms in the hot springs. The presence of ions may contribute to the complex range of colors in microbial mats (Boidi et al., 2022); this effect was observed in the microbial mats studied in the TG where various colors such as white, gray, yellow, red, pink, and green were observed, which coincides with the presence of sulfate ions, and other metals like Fe, Cd, Cu, Cr, and Al among others.
External factors can also influence the color of microbial mats. Dunckel et al. (2009) observed that temperature has a marked effect on the mats’ color and the pigments of the microorganisms, mainly phototrophs, whose pigments absorb at different wavelengths. On the other hand, Bachar et al. (2008) found that metabolism also influences the color of microbial mats; for example, Chloroflexus auriantacus can present 2 colorations depending on the environment where it grows phototropically, and in anaerobic conditions, it shows green-yellow coloration while when it grows chemoheterotrophically in aerobic conditions, it gives red-orange colorations. It follows that the color of microbial mats depends on various factors, both environmental (temperature, pH, and chemical composition of the water) and metabolic (aerobic and anaerobic conditions and phototrophic or chemoheterotrophic metabolism) that, in combination, produce a wide range of colors.
In the TG, a great diversity of thermophilic microorganisms was detected. The phylum Armatimonadetes was the most abundant in the GD sample, whose temperature at the sampling time was 62 °C. This phylum was found in hot springs in different parts of the world with temperatures close to TG, as in the case of Tibetan hot springs (Xian et al., 2020; Zhang et al., 2018), Sungai Klah hot springs, in Malaysia (Chan et al., 2017), Great Boiling Spring (Cole et al., 2013), and in Octopus Springs in Yellowstone National Park, both in the USA (Im et al., 2012).
The class Chthonomonadetes is part of this phylum, which is present in the GD sample. The only strain of this class isolated and cultivated is Chthonomonas calidirosea, which grows at 50-73 °C (Lee et al., 2011).
Cyanobacteria are vital in biogeochemical and photosynthetic processes; they reduce N2 to ammonium. In light conditions and aerobic respiration, it generates large amounts of energy, switching to fermentative metabolism at night when microbial mats become anoxic. Given its importance, it is common to find Cyanobacteria in various environmental conditions, Garga, Russia, in Camargo wetlands, southern France, in thermal areas of the Pakistanis Himalayas, in the Colombian Andes (El Coquito), Yellowstone Park (Bennett et al., 2020), and in Ghats, India (Amin et al., 2017; Berlanga et al., 2017; Rozanov et al., 2017; Subudhi et al., 2017; Yim et al., 2006).
Another abundant phylum in the TG was Chloroflexi. its presence predominates in hot springs with temperatures of 53-65 °C and slightly alkaline pH (7.75-7.91) (Chiriac et al., 2017), which are conditions like those found in the TG waters. Chloroflexi has been detected in microbial mats in hot springs of Patagonia (Alcamán-Arias et al., 2018), Costa Rica (Uribe-Lorío et al., 2019), Japan (Hanada et al., 2002), Yellowstone (USA) (Spieck et al., 2020), Kamchatka, Thailand, Tibet (Saw et al., 2013), and the Andes (Delgado-Serrano et al., 2014).
Another factor that determines the microbial mat communities in the hot springs is temperature. In the case of the class Chloroflexia, temperature has a marked effect on the presence of microorganisms. It has been observed that the family Roseiflexineae prevails at 60 °C, while at 70 °C, the family Chloroflexineae is predominant (Bennett et al., 2020; Hanada et al., 2002). These observations were corroborated in the TG waters, where the family Roseiflexineae was the most abundant below 62 °C (GB and GC samples). In contrast, the family Chloroflexineae was more abundant in the GA sample, which presented a temperature of 94 °C at the sampling time.
The phylum Deinococcus-Thermus includes the class Deinococci that was found in the TG. These bacteria and their cellular components are of biotechnological interest for their possible application in bioremediation (Albuquerque & da Costa, 2014). The phylum Deinococcus-Thermus is a group of microorganisms abundant in hot springs (Kadnikov et al., 2021; Rosenberg, 2014). This phylum was described in hot springs and microbial mats of Tibet (China), Sichuan (China), Yellowstone (USA), Siloam (South Africa), Bulgaria (Eastern Europe), and Chile (South America) (Alcamán-Arias et al., 2018; Zhang et al., 2018). This phylum plays a very important role in carbon, nitrogen, and sulfur cycles, such as Thermus oshimai, T. thermophilus (Murugapiran et al., 2013), or Thermus scotoductus with oxidizing mixotrophic characteristics of sulfur (Skirnisdottir et al., 2014). In TG, the genus Thermus is possibly responsible for taking advantage of the sulfur compounds in the TG water.
Another abundant phylum in the TG samples was the Proteobacteria. They have also been found in microbial mats, sediments, and hot springs of Eritrea (Africa), with relative abundances of 6.2 to 82.3% at temperatures of 44 to 110 °C and pH of 6.97 to 7.54 (Ghilamicael et al., 2017). Still, its presence has also been reported in habitats with temperatures of 29-35 °C and pH 3.5-6.5, which decreases significantly at 68 °C, and pH 6.9 (Delgado-Serrano et al., 2014). This allows deducing that phylum Proteobacteria inhabit a wide variety of environmental conditions.
Another phylum found in the TG was Bacteroidetes, which is a group of anaerobic and gram-negative chemoautotrophic organisms that do not form endospores and are not mobile by sliding, with wide distribution in the environment (Krieg et al., 2011), with presence in water samples, wet sediments, and microbial mats from hot springs from Eritrea, Ethiopia (49.5 °C -100 °C, pH of 6.97-7.54), the Himalayas (60-80 °C; pH 8.0-8.5) (Gupta et al., 2017), and Baikal Rift zone (Stom et al., 2022), where similar conditions to those measured in the TG are found.

Analysis of other identified taxonomic groups
The phyla Acidobacteria, Actinobacteria, Aquificae, Chlorobi, Dictyoglomi, Firmicutes, Gemmatimonadetes, Nitrospirae, Planctomycetes, Spirochaetes, Synergistetes, Tenericutes, Thermotogae, and Verrucomicrobia were identified, those phyla have been reported for their ability to grow in extreme temperature and alkaline environments (Chan et al., 2017).
Bacteria of the phylum Arquea were also detected, including Euyarchaeota and Crenarchaeota. The phylum Euryarchaeota includes halophilic, methanogenic, thermoacidophilic, and hyperthermophilic. These bacteria have been found in the Lake Baikal rupture, where archaea, mainly Crenarchaeota, constituted 19.8% of the total number of sequences in areas with a temperature of 74 °C (Ma et al., 2021; Rozanov et al., 2017). These results agree with the results obtained in this work; it was observed that the elevated temperature favors the growth of Crenarchaeota because, in the sampling area, the temperature was between 62 and 65 °C, very close to the optimal growth temperature reported by other authors.
Alpha diversity analysis. In the 4 samples of the TG, differences were observed between them; the GD sample of light blue color presented the greatest diversity because it does not show a defined asymptote (Fig. 3A). Similar behavior is observed among the remaining samples. They follow an asymptotic trend, suggesting that the greatest diversity possible was identified. The Good coverage graph measures how well the sampling was conducted in an environment (Fig. 3B). It indicates the percentage of individuals sampled in a microbial community, where values greater than 0.9%, evidence that the sequencing effort is sufficient to represent the largest number of species (Lemos et al., 2011). The result observed in Figure 3B showed that the sequencing effort was sufficient to represent the largest number of species. Regarding the observed richness, the Chao1 index was calculated considering the total number of species (Fig. 3C), rare species, and those found twice in the sample (Feng et al., 2021). The results indicated greater richness in the GD sample, followed by GA, GB, and GC. The Shannon-Weiner index (diversity) was defined as a species richness and uniformity estimator (Malvandi et al., 2021; Fig. 3D). Typical values are generally between 1.5 and 3.5 in most ecological studies, and the index rarely exceeds 4 (Feghali et al., 2019; Kim et al., 2017). The results reflect that the GD (microbial mat-sediments) and GC (microbial mat II) samples have greater diversity because both exceed the value of 3.5. Still, the GD sample is even more abundant because it is ~ 5; in addition, values greater than 4 have greater diversity and are not so common. In the case of the GA and GB samples, they have values of around 3, which indicate that they have an average diversity.
The Simpson metric, also called dominance index (Fig. 3E), gives a higher weight to common or similar species and underestimates rare species (Magurran, 2004). Therefore, in the GB sample, there is a greater probability of predominance of a particular species, followed by the GD sample. Finally, the GA and GC samples presented similar percentages.
The results obtained from the beta diversity indicated a great microbial diversity in the 4 samples analyzed, since most OTU´s were not shared. Temperature can explain this result; the GA sample was taken from the geyser source at 94 ºC, while for the GB, GC, and GD samples, the temperatures were 65, 61.5, and 62 ºC, respectively. Similar results have been obtained from other hot spring areas, where it has been observed that microbial diversity depends on the sampling area (Giguète & Tunnicliffe, 2021). Central microbiome analysis is used to understand stable and consistent components across complex microbial ensembles. The results were obtained according to the “affiliation” technique based on the presence and absence of OTU´s between the different microbiomes that are compared, considering 1,623 bp for the normalization of the data (rarefaction). It was observed that the 4 samples are very diverse due to the temperature of the sampling, which differentiates the populations.
Based on the obtained results, the importance of ATP-dependent transporters or ABC transporters that belong to the class of primary transporters is deduced as being very important routes in archaea and bacteria. In the case of extremophiles, these transporters are used as a survival mechanism to regulate osmotic pressure and the generation of exopolysaccharides. They are part of one of the main transport routes at high temperatures. In archaea, they can accumulate substrates at much higher concentrations within cells (Koning et al., 2002). Kawai et al. (2021), when analyzing the metabolic versatility of Chloroflexus aggregans, determined that photosynthetic metabolism, oxidation-reduction, carbon fixation, and transport through the membrane are the most important and contribute to the survival of the microorganism. Within the communities detected in the TG, the presence of the phylum Chloroflexi is observed in all the samples. Hence, the results obtained from the analysis of the functional profile coincide with those described by this author. On the other hand, Jasser et al. (2022) studied the factors that contribute to the adaptability of Cyanobacteria; they observed that the metabolism involved in the synthesis of genes that encode heat shock proteins as well as other proteins such as polyketide synthases influence the adaptability of these microorganisms at high temperatures. In the results obtained from the analysis of the TG samples, this type of metabolism was also detected, in addition to the fact that the species Cyanobacteria were detected in the 4 samples studied.
DNA repair and the maintenance of the genome and its expression is an established mechanism due to growth at high temperatures, where DNA decomposition is accelerated. Some repair proteins are widely conserved, observed mainly in hyperthermophilic archaea. In addition, under high radiation levels, including ionizing and UV radiation, repair of massive DNA damage is mediated by energy and protein metabolism, occurring mainly in thermophilic organisms (Krisko & Radman, 2013). Wang et al. (2023) confirmed the existence of highly efficient DNA repair systems in Cyanobacteria and Chloroflexi, which allows them to survive in hot springs; these phyla were also found in the TG samples analyzed, and through functional analysis of metabolic profiles allows confirming the presence of this type of metabolism.
Regarding energy metabolism, the results agree with the relative abundances found for some bacterial groups where aerobic respiration is the main mechanism for energy generation. In this case, oxidative phosphorylation indicates that electron transfer to a terminal electron receptor, such as oxygen, nitrate, or sulfate, generates ATP. Similar results have been reported in hot springs in Finland (Bomberg et al., 2016). This shows that thermal environments follow a trend in predicting metabolic functions. However, some of them showed results associated with activities in thermal environments; in others, the metabolic capacities of the microbial community were not fully reflected, as is the case of sulfur metabolism, which is very reduced; this can be attributed to a low quantity and quality of the genomes annotated in the databases that are related to the species observed in the thermal water samples (Sahoo et al., 2016). In this research, the microbial communities present in the microbial mats, sediments, and saline accumulations were described for the first time in the thermophilic environment of the TG, Hidalgo, Mexico. Thirty-two phyla were identified of which: Armatimonadetes, Chloroflexi, Cyanobacteria, Thermi, Proteobacteria, Bacteroidetes, Spirochaetes, Firmicutes, Acidobacteria, Nitrospirae, Crenarchaeota, and Euryarchaeota, were the most abundant. This demonstrates the great diversity and abundance of microorganisms found in the geyser, which depend on the continuous supply of nutrients from inorganic reduced sulfur compounds, elevated temperatures, and continuous exposure to light, resulting in phototrophic microbial mats.
The functional predictions of the PiCRUSt-KEGG profiles indicated metabolic pathways related to membrane transport, carbohydrate metabolism, amino acid metabolism, energy metabolism, replication and repair, cofactor, and vitamin metabolism. Membrane transport is widely related to the ABC transporters, the phosphotransferase system, and the bacterial secretion system, which contribute substantially to the adaptability of these microorganisms to thermal environments, to name a few. The results of this research provided new prospects for the industrial, scientific, and biotechnological potential that can be derived from thermophilic microorganisms.
Acknowledgments
The present work was funded by the National Council of Science and Technology (Conacyt), Mexico.
References
Albuquerque, L., & da Costa, M. S. (2014). The Family Thermaceae. In E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, & F. Thompson (Eds.), The Prokaryotes (pp. 955–987). Berlin: Springer. https://doi.org/10.1007/978-3-642-38954-2_128
Alcamán-Arias, M. E., Pedrós-Alió, C., Tamames, J., Fernández, C., Pérez-Pantoja, D., Vásquez, M. et al. (2018). Diurnal changes in active carbon and nitrogen pathways along the temperature gradient in porcelana hot spring microbial mat. Frontiers in Microbiology, 9, 1–17. https://doi.org/10.3389/fmicb.2018.02353
Almeida, A., Mitchell, A. L., Tarkowska, A., & Finn, R. D. (2018). Benchmarking taxonomic assignments based on 16S rRNA gene profiling of the microbiota from commonly sampled environments. Gigascience, 7, 1–10. https://doi.org/10.1093/gigascience/giy054
Amin, A., Ahmed, I., Salam, N., Kim, B., Singh, D., Zhi, X. et al. (2017). Diversity and distribution of thermophilic bacteria in hot springs of Pakistan. Microbial Ecology, 74, 116–127. https://doi.org/10.1007/s00248-017-0930-1
Atanasova, N., Stoitsova, S., Paunova-Krasteva, T., & Kambourova, M. (2021). Plastic degradation by extremophilic bacteria. International Journal of Molecular Sciences, 22, 5610. https://doi.org/10.3390/ijms22115610
Bachar, A., Polerecky, L., Fischer, J. P., Vamvakopoulos, K., de Beer, D., & Jonkerz, H. M. (2008). Two-dimensional mapping of photopigment distribution and activity of Chloroflexus-like bacteria in a hypersaline microbial mat, FEMS Microbiology Ecology, 65, 434–448. https://doi.org/10.1111/j.1574-6941.2008.00534.x
Bennett, A. C., Murugapiran, S. K., & Hamilton, T. L. (2020). Temperature impacts community structure and function of phototrophic Chloroflexi and Cyanobacteria in two alkaline hot springs in Yellowstone National Park. Environmental Microbiology Reports, 12, 503–513. https://doi.org/10.1111/1758-2229.12863
Berlanga, M., Palau, M., & Guerrero, R. (2017). Functional stability and community dynamics during spring and autumn seasons over 3 years in Camargue microbial mats. Frontiers in Microbiology, 8, 1–13. https://doi.org/10.3389/fmicb.2017.02619
Boidi, F. J., Mlewski, E. C., Fernández, G. C., Flores, M. R., Gérard, E., Farías, M. E. et al. (2022). Community vertical composition of the Laguna Negra hypersaline microbial mat, Puna Region (Argentinean Andes). Biology, 11, 831. https://doi.org/10.3390/biology11060831
Bomberg, M., Lamminmäki, T., & Itävaara, M. (2016). Microbial communities and their predicted metabolic characteristics in deep fracture groundwaters of the crystalline bedrock at Olkiluoto, Finland. Biogeosciences, 13, 6031–6047. https://doi.org/10.5194/bg-13-6031-2016
Canganella, F., & Wiegel, J. (2011). Extremophiles from abyssal to terrestrial ecosystems and possibly beyond. Naturwissenschaften, 98, 253–279. https://doi.org/10.1007/s00114-011-0775-2
Chan, C. S., Chan, K. E. R., Hong, K., Urbieta, M. S., Donati, E. R., Shamsir, M. S. et al. (2017). Effects of physiochemical factors on prokaryotic biodiversity in Malaysian circumneutral hot springs. Frontiers in Microbiology, 1252, 1–14. https://doi.org/10.3389/fmicb.2017.01252
Chiriac, C. M., Szekeres, E., Rudi, K., Baricz, A., Hegedus, A., Dragoş, N. et al. (2017). Differences in temperature and water chemistry shape distinct diversity patterns in thermophilic microbial communities. Applied and Environmental Microbiology, 83, e01363-17. https://doi.org/10.1128/AEM.01363-17
Cole, J. K., Peacock, J. P., Dodsworth J. A., Williams, J. A., Thompson, D. B., Dong, H. et al. (2013). Sediment microbial communities in great boiling spring are controlled by temperature and distinct from water communities. ISME Journal, 7, 718–729. https://doi.org/10.1038/ismej.2012.157
Costa, O. Y. A., Raaijmakers, J. M., & Kuramae, E. E. (2018). Microbial extracellular polymeric substances: ecological function and impact on soil aggregation. Frontiers in Microbiology, 9, 1636. https://doi.org/10.3389/fmicb.2018.01636
Delgado-Serrano, L., López, G., Bohorquez, L. C., Bustos, J. R., Rubiano, C., Osorio-Forero, C. et al. (2014). Neotropical Andes hot springs harbor diverse and distinct planktonic microbial communities. FEMS Microbiology Ecology, 89, 56–66. https://doi.org/10.1111/1574-6941.12333
Dunckel, A. E., Cardenas, M. B., Sawyer, A. H., & Bennett, P. C. (2009). High-resolution in-situ thermal imaging of microbial mats at El Tatio Geyser, Chile shows coupling between community color and temperature. Geophysical Research Letters, 36, L23403. https://doi.org/10.1029/2009GL041366
Feghali, N., Albertin, W., Tabet, E., Rizk, Z., Bianco, A., Zara, G. et al. (2019). Genetic and phenotypic characterisation of a Saccharomyces cerevisiae population of ‘Merwah’ white wine. Microorganisms, 7, 492. https://doi.org/10.3390/microorganisms7110492
Feng, J., Wang, B., Zhang, D., Chu, S., Zhi, Y., Hayat, K. et al. (2021). Streptomyces griseorubens JSD-1 promotes rice straw composting efficiency in industrial-scale fermenter: Evaluation of change in physicochemical properties and microbial community. Bioresource Technology, 321, 124465. https://doi.org/10.1016/j.biortech.2020.124465
Ghilamicael, A. M., Budambula, N. L. M., Anami, S. E., Mehari, T., & Boga, H. I. (2017). Evaluation of prokaryotic diversity of five hot springs in Eritrea. BMC Microbiology, 17, 1–13. https://doi.org/10.1186/s12866-017-1113-4
Giguère, T. N., & Tunnicliffe, V. (2021). Beta diversity differs among hydrothermal vent systems: Implications for conservation. Plos One, 16, e0256637. https://doi.org/10.1371/journal.pone.0256637
Gupta, V., Gupta, N., Capalash, N., & Sharma, P. (2017). Bio-prospecting bacterial diversity of hot springs in northern Himalayan region of India for laccases. Indian Journal of Microbiology, 57, 285–291. https://doi.org/10.1007/s12088-017-0656-2
Hanada, S., Takaichi, S., Matsuura, K., & Nakamura, K. (2002). Roseiflexus castenholzii gen. nov., sp. nov., a thermophilic, filamentous, photosynthetic bacterium that lacks chlorosomes. International Journal of Systematic and Evolutionary Microbiology, 52, 187–193. https://doi.org/10.1099/00207713-52-1-187
Hiriart, G., Gutiérrez-Negrín, L. C. A., Quijano-León, J. L., Ornelas-Celis, A., Espíndola, S., & Hernández, I. (2011). Evaluación de la energía geotérmica en México. Inter-American Development Bank and Comisión Reguladora de Energía. No. 29. Recuperado el 17 septiembre, 2022 de: https://gc.scalahed.com/recursos/files/r161r/w25421w/evaluaciondelaenergia.pdf
Im, W. T., Hu, Z. Y., Kim, K. H., Rhee, S. K., Meng, H., Lee, S. T. et al. (2012). Description of Fimbriimonas ginsengisoli gen. nov., sp. nov. within the Fimbriimonadia class nov., of the phylum Armatimonadetes. Antonie Van Leeuwenhoek, 102, 307–317. https://doi.org/10.1007/s10482-012-9739-6
Jasser, I., Panou, M., Khomutovska, N., Sandzewicz, M., Panteris, E., Niyatbekov, T. et al. (2022). Cyanobacteria in hot pursuit: Characterization of cyanobacteria strains, including novel taxa, isolated from geothermal habitats from different ecoregions of the world. Molecular Phylogenetics and Evolution, 170, 107454. https://doi.org/10.1016/j.ympev.2022.107454
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Karnachuk, O. V., & Ravin, N. V. (2021). Metagenomic analysis of the microbial community in the underground coal fire area (Kemerovo Region, Russia) revealed predominance of thermophilic members of the phyla Deinococcus-Thermus, Aquificae, and Firmicutes. Microbiology, 90, 578–587. https://doi.org/10.1134/S0026261721050088
Kawai, S., Martinez, J. N., Lichtenberg, M., Trampe, E., Kühl, M., Tank, M. et al. (2021). In-situ metatranscriptomic analyses reveal the metabolic flexibility of the thermophilic anoxygenic photosynthetic bacterium Chloroflexus aggregans in a hot spring cyanobacteria-dominated microbial mat. Microorganisms, 9, 652. https://doi.org/10.3390/microorganisms9030652
Kim, B. R., Shin, J., Guevarra, R. B., Lee, J. H., Kim, D. W., Seo, K. H. et al. (2017). Deciphering diversity indices for a better understanding of microbial communities. Journal of Microbiology and Biotechnology, 27, 2089–2093. https://doi.org/10.4014/jmb.1709.09027
Koning, S. M., Albers, S., Konings, W. N., & Driessen, A. J. M. (2002). Sugar transport in (hyper)thermophilic archaea. Research in Microbiology, 2, 61–67. https://doi.org/10.1016/S0923-2508(01)01289-X
Krieg, N. R., Ludwig, W., & Euzéby, J. (2011). Phylum XIV. Bacteroidetes phyl. nov. In N. R. Krieg, J. R. Staley, D. R. Brown, B. P. Hedlund, B. J. Paster, N. L. Ward et al. (Eds.), The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes. New York: Springer. https://doi.org/10.1007/978-0-387-68572-4
Krisko, A., & Radman, M. (2013). Biology of extreme radiation resistance: the way of Deinococcus radiodurans. Cold Spring Harbor Perspectives in Biology, 5, 1–12. https://doi.org/10.1101/cshperspect.a012765
Lee, K. C. Y., Dunfield, P. F., Morgan, X. C., Crowe, M. A., Houghton, K. M., Vyssotski, M. et al. (2011). Chthonomonas calidirosea gen. nov., sp. nov., anaerobic, pigmented, thermophilic micro-organism of a novel bacterial class, Chthonomonadetes classis nov., of the newly described phylum Armatimonadetes originally designated candidate division OP1. International Journal of Systematic and Evolutionary Microbiology, 61, 2482–2490. https://doi.org/10.1099/ijs.0.027235-0
Lemos, L. N., Fulthorpe, R. R., Triplett, E. W., & Roesch, L. F. (2011). Rethinking microbial diversity analysis in the high throughput sequencing era. Journal of Microbiological Methods, 86, 42–51. https://doi.org/10.1016/j.mimet.2011.
03.014
Ma, L., She, W., Wu, G., Yang, J., Phurbu, D., & Jiang, H. (2021). Influence of temperature and sulfate concentration on the sulfate/sulfite reduction prokaryotic communities in the tibetan hot springs. Microorganisms, 9, 583. https://doi.org/10.3390/microorganisms9030583
Malvandi, H., Moghanizade, R., & Abdoli, A. (2021). The use of biological indices and diversity indices to evaluate water quality of rivers in Mashhad, Iran. Biologia, 76, 959–971. https://doi.org/10.2478/s11756-020-00618-4
Magurran, A. E. (2004). Measuring biological diversity. Current Microbiology, 31, R1174–R1177. https://doi.org/10.1016/j.cub.2021.07.049
Mackenzie, R., Pedrós-Alió, C., & Díez, B. (2013). Bacterial composition of microbial mats in hot springs in Northern Patagonia: variations with seasons and temperature. Extremophiles, 17, 123-136. https://doi.org/10.1007/s00792-
012-0499-z
Murugapiran, S. K., Huntemann, M., Wei, C. L., Han, J., Detter, J. C., Han, C. et al. (2103). Thermus oshimai JL-2 and T. thermophilus JL-18 genome analysis illuminates pathways for carbon, nitrogen, and sulfur cycling. Standards in Genomic Sciences, 7, 449–68. https://doi.org/10.4056/sigs.3667269
Nicolaus, B., Kambourova, M., & Oner, E. T. (2010). Exopolysaccharides from extremophiles: from fundamentals to biotechnology. Environmental Technology, 10, 1145–1158. https://doi.org/10.1080/09593330903552094
Nimick, D. A., Moore, J. N., Dalby, C. E., & Savka, M. W. (1998). The fate of geothermal arsenic in the Madison and Missouri rivers, Montana and Wyoming. Water Resources Research, 34, 3051–3067. https://doi.org/10.1029/98WR01704
Nuñez-Benítez, J. O. (2007). Caracterización hidrogeoquímica de las aguas subterráneas y superficiales del municipio de Tecozautla, Estado de Hidalgo (Ph. D. Thesis). Universidad Autónoma del Estado de Hidalgo. Pachuca, México.
Prieto-Barajas, C. M., Alfaro-Cuevas, R., Valencia-Cantero, E., & Santoyo, G. (2017). Effect of seasonality and physicochemical parameters on bacterial communities in two hot spring microbial mats from Araró, Mexico. Revista Mexicana de Biodiversidad, 88, 616–624. https://doi.org/10.1016/j.rmb.2017.07.010
Reed, J. C., Lewis, H., Trejo, E., Winston, V., & Evilia, C. (2013). Protein adaptations in Archaeal extremophiles. Archea, 213, 1–14. https://doi.org/10.1155/2013/373275
Rinehart, J. S. (1980). Geysers and geothermal energy. New York: Springer-Verlag. https://doi.org/10.1007/978-1-4612-6084-4
Rosenberg, E. (2014). The Family Deinococcaceae. In E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, & F. Thompson (Eds.), The Prokaryotes (pp. 613–615). Berlín: Springer. https://doi.org/10.1007/978-3-642-38954-2_127
Rozanov, A. S., Bryanskaya, A. V., & Ivanisenko, T. V. (2017). Biodiversity of the microbial mat of the Garga hot spring. BMC Evolutionary Biology, 17, 38–64. https://doi.org/10.1186/s12862-017-1106-9
Rudi, K., Skulberg, O. M., Larsen, F., & Jakobsen, K. S. (1997). Strain characterization and classification of oxyphotobacteria in clone cultures on the basis of 16S rRNA sequences from the variable regions V6, V7 and V8. Applied and Environmental Microbiology, 63, 2593–2599. https://doi.org/10.1128/AEM.63.7.2593-2599.1997
Sahoo, K. R., Gaur, M., Das, A., Singh, A., Kumar, M., & Subudhi, E. (2016). Comparative analysis of 16S rRNA Gene Illumina Sequence for microbial community structure in diverse unexplored hot springs of Odisha, India. Geomicrobiology Journal, 34, 567–576. https://doi.org/10.1080/01490451.2016.1238980
Saw, J. H. W., Schatz, M., Brown, M. V., Kunkel, D. D., Foster J. M., Shick, H. et al. (2013). Cultivation and complete genome sequencing of Gloeobacter kilaueensis sp. nov., from a Lava Cave in Kilauea Caldera, Hawaii. Plos One, 8, e76376. https://doi.org/10.1371/journal.pone.0076376
Skirnisdottir, S., Hreggvidsson, G., Holst, O., & Kristjansson, J. K. (2014). Isolation and characterization of a mixotrophic sulfur-oxidizing Thermus scotoductus. Extremophiles, 5, 45–51. https://doi.org/10.1007/s007920000172
Spieck, E., Spohn, M., Wendt, K., Bock, E., Shively, J., Frank, J. et al. (2020). Extremophilic nitrite-oxidizing Chloroflexi from Yellowstone hot springs. The ISME Journal, 4, 364–379. https://doi.org/10.1038/s41396-019-0530-9
Stom, D. I., Topchy, I. A., Zhdanova, G. O., Barkhutova, D. D., Zaitseva, S. V., Kupchinsky, A. B. et al. (2022). Microorganisms of microbial mats from an alkaline hot spring of Baikal rift zone as bioagents in a biofuel cell. Geomicrobiology Journal, 39, 566–576. https://doi.org/10.1080/01490451.2022.2054030
Subudhi, E., Sahoo, R. K., Gaur, M., Singh, A., & Aradhana, D. (2017). Shift in Cyanobacteria community diversity in hot springs of India. Geomicrobiology Journal, 35, 141–147. https://doi.org/10.1080/01490451.2017.1338799
Tamazawa, S., Takasaki, K., Tamaki, H., Kamagata, Y., & Hanada, S. (2012). Metagenomic and biochemical characterizations of sulfur oxidation metabolism in uncultured large sausage-shaped bacterium in hot spring microbial mats. Plos One, 7, e49793. https://doi.org/10.1371/journal.pone.0049793
Uribe-Lorío, L., Brenes-Guillén, L., Hernández-Ascencio, W., Mora-Amador, R., González, G., Ramírez-Umaña, C. J. et al. (2019). The influence of temperature and pH on bacterial community composition of microbial mats in hot springs from Costa Rica. Microbiology Open, 8, 1–26. https://doi.org/10.1002/mbo3.893
Wang, X., Yin, Y., Yu, Z., Shen, G., Cheng, H., & Tao, S. (2023). Distinct distribution patterns of the abundant and rare bacteria in high plateau hot spring sediments. Science of The Total Environment, 863, 160832. https://doi.org/10.1016/j.scitotenv.2022.160832
Ward, L. M., Idei, A., Nakagawa, M., Ueno, Y., Fischer, W. W., & McGlynn, S. E. (2019). Geochemical and metagenomic characterization of Jinata Onsen, a Proterozoic-analog hot spring, reveals novel microbial diversity including iron-tolerant phototrophs and thermophilic lithotrophs. Microbes and Environments, 34, 278–292. https://doi.org/10.1264/jsme2.ME19017
Xian, W. D., Salam, N., Li, M. M., Zhou, E. M., Yin, Y. R., Liu, Z. T. et al. (2020). Network-directed efficient isolation of previously uncultivated Chloroflexi and related bacteria in hot spring microbial mats. Biofilms and Microbiomes, 6, 20. https://doi.org/10.1038/s41522-020-0131-4
Yasir, M., Qureshi, A. K., Srinivasan, S., Ullah, R., Bibi, F., Rehan, M. et al. (2020). Domination of filamentous anoxygenic phototrophic bacteria and prediction of metabolic pathways in microbial mats from the hot springs of Al Aridhah. Folia Biology, 66, 24–35.
Yim, L. C., Hongmei, J., Aitchison, J. C., & Pointing, S. B. (2006). Highly diverse community structure in a remote central Tibetan geotermal spring does not display monotonic variation to thermal stress. FEMS Microbiology Ecology, 57, 80–91. https://doi.org/10.1111/j.1574-6941.2006.00104.x
Zhang, Y., Wu, G., Jiang, H., Yang, J., She, W., Khan, I. et al. (2018). Abundant and rare microbial biospheres respond differently to environmental and spatial factors in Tibetan hot springs. Frontiers in Microbiology, 9, 1–16. https://doi.org/10.3389/fmicb.2018.02096


