
Paola Xanath Ruiz-Mendoza a, Jovana M. Jasso-Martínez a, b, *, Jorge Gutiérrez-Rodríguez c, Ernesto Samacá-Sáenz a, d, Alejandro Zaldívar-Riverón a
a Universidad Nacional Autónoma de México, Instituto de Biología, Colección Nacional de Insectos, 3er Circuito Exterior s/n, Cd. Universitaria, Coyoacán, 04510 Ciudad de México, Mexico
b Smithsonian Institution, National Museum of Natural History, Department of Entomology, 10th St. & Constitution Ave. NW, Washington, DC 20560, USA
c Estación Biológica de Doñana, Departamento de Ecología y Evolución, c/Americo Vespucio 26, 41092 Sevilla, Spain
d Universidad Nacional Autónoma de México, Instituto de Investigaciones Biomédicas, Departamento de Biología Celular y Fisiología, 3er Circuito Exterior s/n, Cd. Universitaria, Coyoacán, 04510 Ciudad de México, Mexico
*Corresponding author: jovana.jasso@gmail.com (J.M. Jasso-Martínez)
Received: 1 August 2022; accepted: 15 November 2022
Abstract
The Stenopelmatus talpa species-group (Stenopelmatidae) comprises cricket-like orthopterans distributed across the Trans-Mexican Volcanic Belt (TMVB) morphotectonic province and adjacent areas in central Mexico. Despite recent efforts, the taxonomy and evolutionary relationships for members of this complex still are far from completely known. Here we generated and characterized the mitochondrial (mt) genome of 14 specimens of the S. talpa species-group and evaluated its species limits with the cox1 barcoding locus. Moreover, based on the mt genome DNA sequence data, we also reconstructed its phylogenetic relationships and made inferences about its biogeographic history based on a relaxed molecular clock analysis. A total of 9 species were delimited using a 2% pairwise distance criterion, which were consistent with our best estimate of phylogeny. The relationships recovered for the S. talpa species-group were similar although with more recent divergence time estimates than those obtained in a previous phylogenetic study, suggesting that its origin and subsequent diversification in the TMVB followed an east-central pattern, with its earliest divergence occurring during the late Pliocene to early Pleistocene.
Keywords: Trans-Mexican Volcanic Belt; Ensifera; Mesoamerica; DNA barcoding
© 2023 Universidad Nacional Autónoma de México, Instituto de Biología. This is an open access article under the CC BY-NC-ND license
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Caracterización del genoma mitocondrial y filogenia del complejo Stenopelmatus talpa del centro de México (Orthoptera: Stenopelmatidae: Stenopelmatini)
Resumen
El grupo de especies Stenopelmatus talpa (Stenopelmatidae) agrupa a ortópteros similares en apariencia a grillos que se encuentran distribuidos en la provincia morfotectónica del Eje Neovolcánico Transversal (ENT) y áreas adyacentes en el centro de México. A pesar de esfuerzos recientes, la taxonomía y las relaciones evolutivas de los miembros de este complejo aún están lejos de conocerse por completo. En este trabajo se caracteriza el genoma mitocondrial de 14 ejemplares del grupo de especies S. talpa y se evalúan sus límites de especie utilizando el marcador mitocondrial cox1. Con base en la secuencia completa del genoma mitocondrial se reconstruyeron las relaciones filogenéticas del grupo y se hicieron inferencias sobre su historia biogeográfica. Se delimitaron un total de 9 especies con base en un criterio de 2% de divergencia, las cuales fueron consistentes con nuestra reconstrucción filogenética. Las relaciones obtenidas para el grupo de especies S. talpa fueron similares, aunque con estimaciones de tiempos de divergencia más recientes a las obtenidas en un estudio filogenético anterior, lo que sugiere que su origen y subsecuente diversificación en el ENT siguió un patrón este-centro, ocurriendo el evento de divergencia más temprano entre el Plioceno tardío y el Pleistoceno temprano.
Palabras clave: Eje Neovolcánico Transversal; Ensifera; Mesoamérica; Código de barras de ADN
© 2023 Universidad Nacional Autónoma de México, Instituto de Biología. Este es un artículo Open Access bajo la licencia CC BY-NC-ND
Introduction
The family Stenopelmatidae Burmeister, 1838 is a group of cricket-like orthopterans whose species occur in North and Central America, South and Central Africa, Southeast Asia, and the Indian subcontinent (Cigliano et al., 2022). This family was recently proposed by Gorochov (2021) to contain 3 subfamilies: Stenopelmatinae Burmeister, 1838, Schizodactylinae Blanchard, 1845, and Gryllacridinae Blanchard, 1845. However, only the former subfamily is currently recognized within Stenopelmatidae in the Orthoptera Species File (Cigliano et al., 2022). Following the latter classification, the Stenopelmatinae comprises 5 tribes: Maxentiini Gorochov, 2021, Oryctopini Kevan, 1986, Oryctopterini Gorochov, 1988, Siini Gorochov, 1988, and Stenopelmatini Burmeister, 1838 (Cigliano et al., 2022; Gorochov, 2021). Of these tribes, Stenopelmatini is the only one present in the American continent, with species distributed from British Columbia in Canada along the west coast of the USA to the Mesoamerican highlands from northern Mexico to Panama, and with one species from northern South America in Ecuador (Cigliano et al., 2022; Weissman et al., 2021).
Members of Stenopelmatini, commonly known as Jerusalem crickets in the U.S. and as ‘cara de niño’ or ‘niño de la tierra’ in Mexico, are amongst the most recognizable insects in North and Mesoamerica, (Weissman, 2005). They are mainly nocturnal, hiding under rocks or logs during the day (Weissman, 2001, 2005). Species of this tribe mainly feed on roots and are considered important organic matter decomposers (Weissman, 2001), although they also feed on small invertebrates (Weissman, 2001).
In a molecular phylogenetic study for the Stenopel-
matoidea based on 2 nuclear and 1 mitochondrial (mt) gene markers (Vandergast et al., 2017), the Stenopelmatini was recovered as monophyletic, although, only members of another tribe, Siini, were included. In a more recent integrative phylogenetic study that focused on the Stenopelmatini, Weissman et al. (2021) recovered 3 strongly supported main clades for the subfamily based on both anchor hybrid enrichment and mt data. One of these clades had the micropterous S. piceiventris Walker, 1869 from Oaxaca in southeast Mexico, whereas the remaining 2 clades included species from western North America, Canada to northern Mexico, and Mesoamerica, respectively. Based on these relationships and examination of morphological, behavioral, and chromosomal features, the tribe was restricted to contain only 2 genera: Ammopelmatus Tinkham, 1965 for the North American clade and Stenopelmatus Burmeister, 1838 for the 2 clades with Mesoamerican species.
The genus Stenopelmatus currently contains 19 described macropterous, micropterous, and apterous species that mainly inhabit highlands with pine-oak and cloud forests (Cigliano et al., 2022; Weissman et al., 2021). A recent phylogenomic study showed that the species richness in this genus is considerably higher, with several undescribed species with a marked geographic phylogenetic structure (Gutiérrez-Rodríguez et al., 2022). Based on these results, 4 species-groups were proposed: the faulkneri, talpa, Central American and piceiventris species-groups, whose geographic distribution and molecular clock estimates were concordant with some geological processes that took place in the Mexican Transition zone (Gutiérrez-Rodríguez et al., 2022).
The S. talpa species-group was proposed by Gutiérrez-Rodríguez et al. (2022) to contain 2 described (S. talpa Burmeister, 1838 and S. typhlops Rehn, 1903) and 5 undescribed species that occur across the Trans-Mexican Volcanic Belt (TMVB) and adjacent areas of the Sierra Madre Oriental (SMO) morphotectonic provinces in central Mexico. However, despite the effort of the above cited authors to assess the species boundaries in this group, additional information from different sources is still required to confirm its species limits, phylogenetic relationships, and biogeographic history.
Mt sequence data have been widely employed to elucidate evolutionary relationships, establish species boundaries, and carry out population genetics studies (Avise et al., 1987; Ballard & Pichaud, 2014). The metazoan mt genome typically consists of 15-18 kilobases, comprising 13 protein-coding genes, 22 transfer RNAs (tRNAs), and 2 ribosomal RNAs (rRNAs) (Boore, 1999), with this composition being generally conserved across bilaterian metazoans (Cameron, 2014; Shen et al., 2016). The use of this locus has given access to a set of orthologous genes with little or no recombination (Ballard & Rand, 2005) and has proved to provide valuable phylogenetic resolution at both shallow and deep evolutionary scales (Ballard & Rand, 2005; Cameron, 2014; DeSalle et al., 2017; Lumley & Sperling, 2010; Song et al., 2015).
With the advent of next-generation sequencing (NGS) techniques, phylogenetic studies of a number of biological groups, including insects, have enormously benefited due to the facilitated generation and use of genomic information, including the assembly of complete mt genomes (Maddock et al., 2016). In this respect, it has been shown that phylogenetic analyses of complete mt genome data increase resolution for resolving species diversification events (Bae et al., 2004; Song et al., 2015).
Here we generated and characterized 16 complete and partial mt genomes of Stenopelmatus species, 14 of which belong to species of the S. talpa species-group that were delimited in Gutiérrez-Rodríguez et al.’s (2022) study, and whose validity still needs to be further corroborated. We employed this mt genome data to assess the species limits among the examined taxa, estimate their phylogenetic relationships, and investigate their biogeographical history in central Mexico. Our results support the existence of at least 9 species within the S. talpa species-group, 2 of which have already been described, whose geographic structure is congruent with the geological history of the TMVB and adjacent morphotectonic provinces.
Materials and methods
A total of 16 Stenopelmatus specimens were included in this study, 14 of which belong to the S. talpa species-group. These specimens were collected in localities situated in 7 Mexican states across the TMVB and SMO provinces (Hidalgo, Mexico City, Michoacán, Puebla, Querétaro, State of Mexico, and Veracruz; Fig. 1). Our sampling included specimens assigned to the only 2 described species of the S. talpa species-group, S. talpa and S. typhlops, which were collected from their type locality. We also included 2 undescribed species from 2 other species-groups: one belonging to the faulkneri species-group, S. sp. aff. mineraldelmonte (El Chico, Hidalgo, CNIN3922), and a second to the Central America species-group, rooting the trees with the latter species. A list with the examined species, their locality, DNA voucher, and GenBank accession numbers is provided in Table 1. All samples were collected between the years 2012 to 2018 and preserved in 96% ethanol at -20°C until they were processed for DNA sequencing.
Genomic DNA was extracted from smooth muscle of hind femur with the EZ-10 Spin Kit minipreps DNA Genomic Column Kit (BIOBasic, Toronto, Ontario, Canada) following the manufacturer’s protocol. DNA fragmentation was performed with the Bioruptor Pico equipment (Diagenode Inc.) employing 3 cycles of sonication (15-90 sec. on-off pulse). We used 100 ng of input DNA for library preparation with the Kapa Hyper Prep kit (Kapa Biosystems Inc., Wilmington, MA, U.S.A), and the TruSeq-style dual-indexing adapters (Glenn et al., 2016). Genomic DNA quantitations, pre- and post-PCR libraries, were performed using a Qubit 2.0 fluorometer (Invitrogen, Life Technologies, CA, USA). Whole-genome shotgun sequencing was conducted with an Illumina HiSeq X instrument at the Department of Environmental Health Science, University of Georgia, Athens, GA, USA.
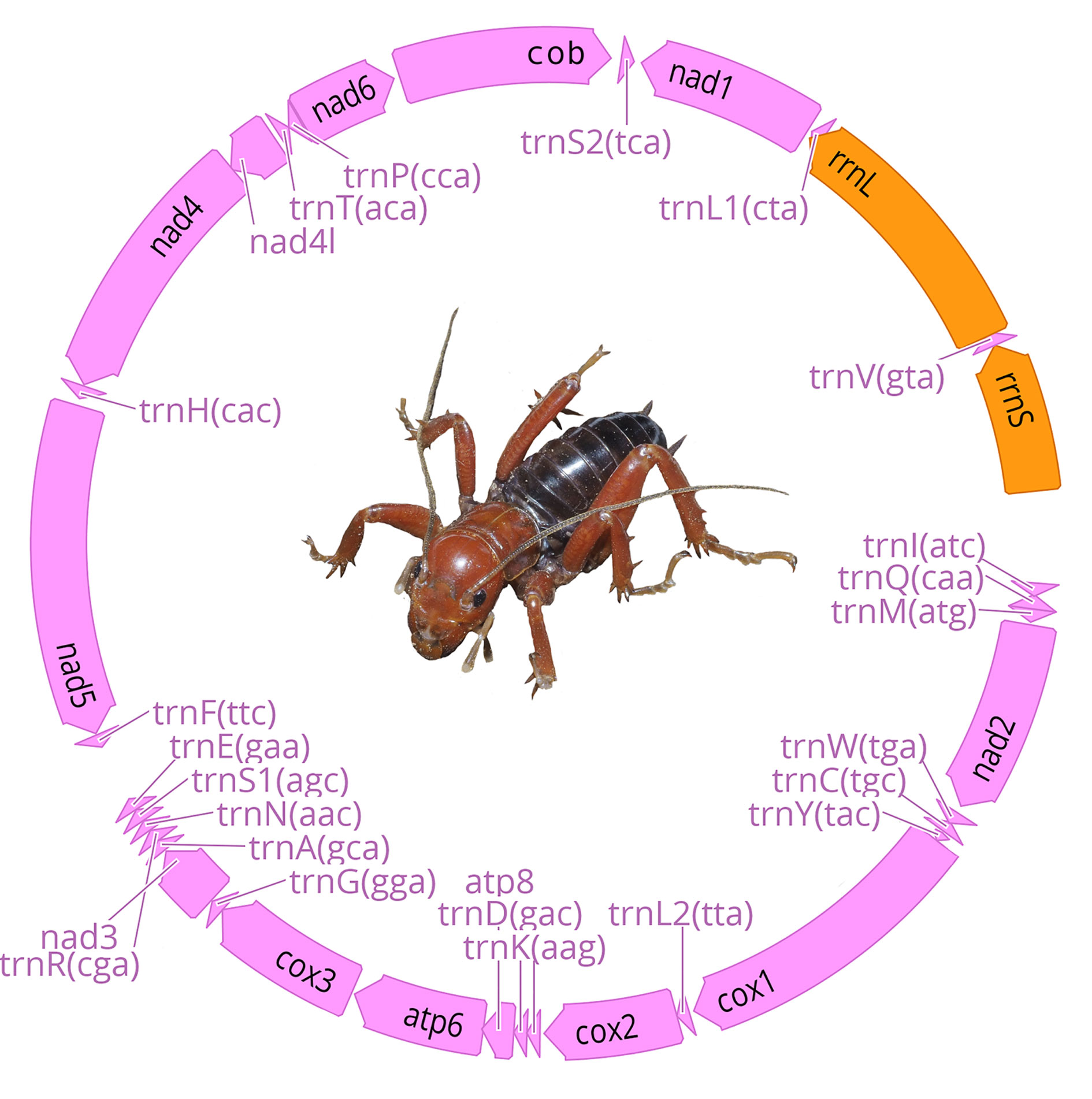
Raw reads were trimmed and filtered for all samples using the program Geneious version 10.2.6 (Kearse et al., 2012). We carried out mt genomes de novo assemblies in the program GetOrganelle (Jin et al., 2018) using the mtDNA database. For 3 samples (CNIN 3669, 3741, 3922) we could not recover complete mt genomes in the de novo assembly. In these cases, we performed by-reference assemblies with Geneious to obtain longer contigs, using as reference either mt sequence genomes of geographically close samples or their cytochrome oxidase I (cox1) sequence previously obtained by Sanger sequencing. Mt genome annotations were performed with the MITOS 2 web server (Bernt et al., 2013) and verified using the protein-coding genes signal from the “protein plots” generated by MITOS. We used Geneious to confirm the accuracy of our assemblies and annotations. We also registered the order of the mt genome genes to identify possible gene arrangements, and then compared them to the Pancrustacea ground pattern, which is the proposed Crustacea/Hexapoda common ancestor (Boore et al., 1995, 1998).
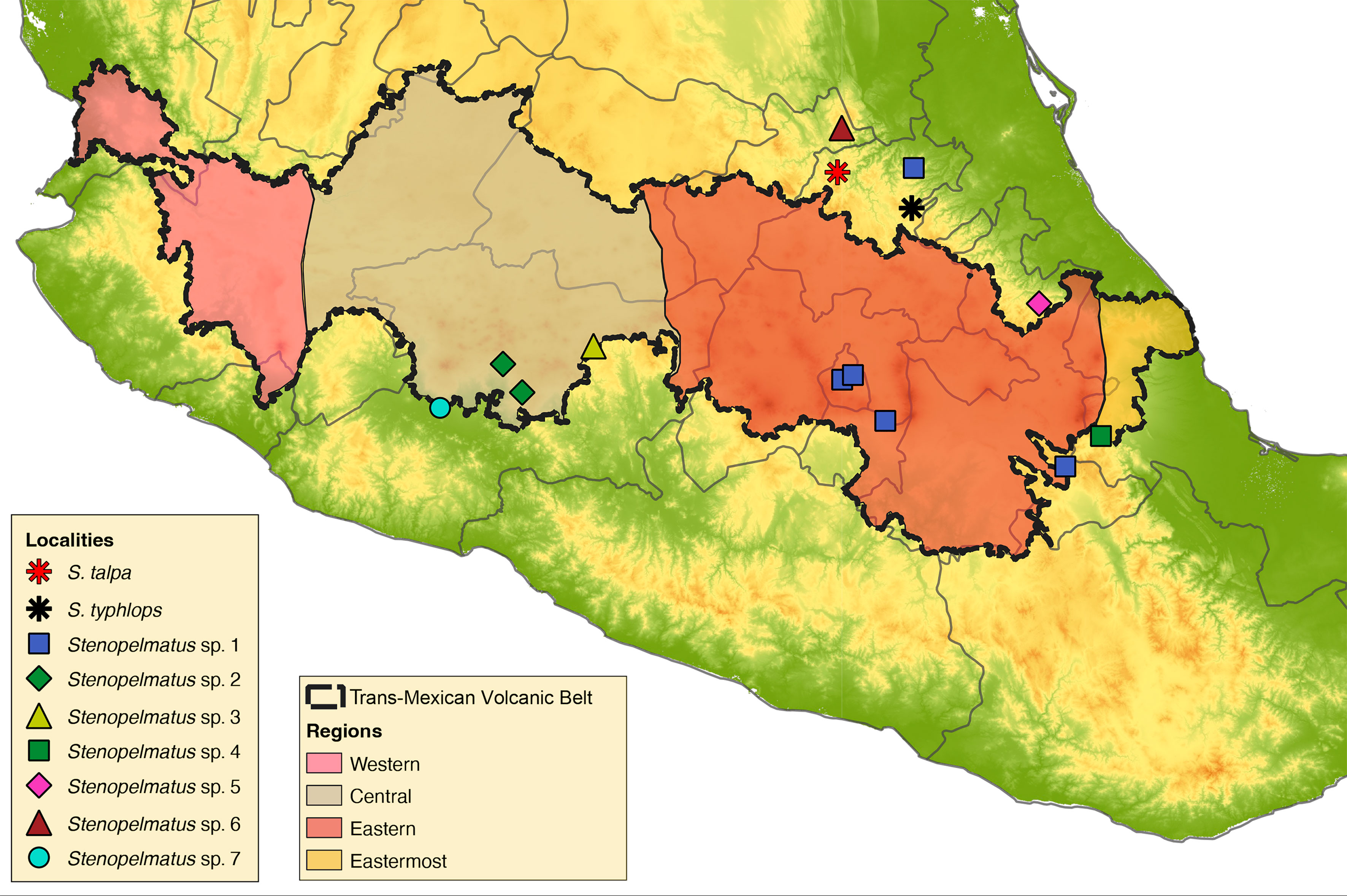
Table 1
Specimens examined in this study, their locality, geographic coordinates, DNA voucher, Sequence Read Archive (SRA) and Mt GenBank accession numbers.
| Species | Locality | Latitude | Longitude | DNA voucher No. | Raw data SRA accession No. | Mt genome Genbank accession No. |
| S. talpa | Mexico: Hidalgo, Jacala de Ledezma | 20.948 | -99.211 | CNIN4350 | SRR18074866 | OP778777 |
| S. typhlops | Mexico: Hidalgo, Zacualtipán | 20.636 | -98.628 | CNIN3983 | SRR18074868 | OP699672 |
| Stenopelmatus sp. 1 | Mexico: Estado de México, Nepantla | 18.980 | -98.833 | CNIN3651 | SRR18074864 | OP778769 |
| Stenopelmatus sp. 1 | Mexico: CDMX, Coyoacán | 19.306 | -99.174 | CNIN3667 | SRR18074875 | OP778770 |
| Stenopelmatus sp. 1 | Mexico: CDMX, Iztapalapa | 19.343 | -99.089 | CNIN3669 | SRR18074869 | OP778771 |
| Stenopelmatus sp. 1 | Mexico: Puebla, Chapulco | 18.619 | -97.410 | CNIN3989 | SRR18074862 | OP778775 |
| Stenopelmatus sp. 1 | Mexico: Hidalgo, Tlanchinol | – | – | CNIN4152 | SRR18074861 | OP699671 |
| Stenopelmatus sp. 2 | Mexico: Michoacán, Ario de Rosales | 19.205 | -101.704 | CNIN3934 | SRR18074872 | OP778774 |
| Stenopelmatus sp. 2 | Mexico: Michoacán, Tingambato | 19.430 | -101.856 | CNIN3736 | SRR18074874 | OP778772 |
| Stenopelmatus sp. 3 | Mexico: Michoacán, Morelia | 19.572 | -101.140 | CNIN3737 | SRR18074863 | OP699673 |
| Stenopelmatus sp. 4 | Mexico: Veracruz, Orizaba | 18.860 | -97.128 | CNIN3639 | SRR18074873 | OP778767 |
| Stenopelmatus sp. 5 | Mexico: Puebla, Zacapoaxtla | 19.908 | -97.617 | CNIN3662 | SRR18074876 | OP699674 |
| Stenopelmatus sp. 6 | Mexico: Querétaro, Valle de Guadalupe | 21.299 | -99.177 | CNIN3646 | SRR18074867 | OP778768 |
| Stenopelmatus sp. 7 | Mexico: Michoacán, Apatzingán | 19.083 | -102.353 | CNIN3741 | SRR18074870 | OP429107 |
| Stenopelmatus sp. aff. mineraldelmonte (faulkneri species-group) | Mexico: Hidalgo, El Chico | 20.184 | -98.716 | CNIN3922 | SRR18074871 | OP778773 |
| Stenopelmatus sp. (Central America species-group) | Costa Rica: San José, Rivas Chirripó | 9.464 | -83.577 | CNIN4267 | SRR18074865 | OP778776 |
We extracted the sequences of the 13 protein-coding genes from all samples and independently aligned them using the program MAFFT version 7 (Katoh & Standley, 2013). We then verified each protein-coding gene alignment with respect to the reading frame (invertebrate mt genetic code) in Geneious. The 2 ribosomal genes (rRNA) and the 22 transfer genes (tRNA) from each sample were also extracted and manually aligned. The following 3 matrices were obtained: 1) a cox1 matrix comprising the DNA barcoding locus (658 bp), 2) a protein-coding gene matrix (10,953 bp), and 3) a complete mt genome matrix (14,599 bp). The 3 matrices are available in figshare (https://doi.org/10.6084/m9.figshare.21506583).
Uncorrected genetic distances were calculated both for the complete mitogenome and cox1 matrices with the program PAUP version 4.0a (Swofford, 2002). Mt species delimitation among the samples was assessed for the cox1 data set following a 2% pairwise distance criterion. The use of this marker has been proven to be an efficient tool for species identification of most animal taxa (Hajibabaei et al., 2007; Hebert et al., 2003). We included in this analysis 4 previously published cox1 sequences belonging to specimens of the talpa species-group: 3 from the state of Morelos and one from Mexico City (Gutiérrez-Rodríguez et al., 2022). The uncorrected cox1 distances were visualized building a neighbor-joining (NJ; Saitou & Nei, 1987) tree with PAUP.
Table 2
Main features of the assembled mt genomes in this study: assembly size (bp), mean coverage, and genes not found.
| DNA voucher | Assembly size | GC (%) | Mean coverage | Mt genes not recovered |
| CNIN3662 | 15,270 | 25.6 | 18.3 | – |
| CNIN3667 | 14,249 | 25.4 | 19.7 | – |
| CNIN3983 | 15,806 | 25.3 | 11.4 | – |
| CNIN3646 | 15,440 | 25.0 | 7.9 | – |
| CNIN4350 | 13,558 | 25.2 | 24.7 | trnN, trnS1, trnE, trnF, nad4L, trnT, trnP, trnQ |
| CNIN4267 | 15,485 | 26.1 | 15.6 | – |
| CNIN3651 | 13,485 | 25.9 | 14.5 | trnI, trnA, trnR, trnN, trnS1, trnE, rrnS, trnV, trnF, trnQ |
| CNIN3737 | 15,312 | 25.2 | 15.0 | – |
| CNIN3989 | 14,146 | 25.3 | 13.2 | trnV, rrnS |
| CNIN4152 | 15,483 | 32.3 | 20.5 | – |
| CNIN3736 | 15,356 | 25.1 | 134.1 | trnN, trnR, trnS1 |
| CNIN3639 | 13,056 | 26.5 | 15.3 | trnI, trnQ, trnM, trnV, rrnS |
| CNIN3934 | 13,315 | 25.6 | 5.2 | trnE, rrnL |
| CNIN3922 | 15,785 | 27.5 | 12.9 | trnE |
| CNIN3741 | 14,734 | 24.5 | 16.4 | trnA, trnD, trnF, trnL1, trnR, trnY, rrnL |
| CNIN3669 | 14,860 | 25.2 | 14.8 | trnN, trnS1 |
The best-fit partitioning scheme for the protein-coding gene and complete mt genome matrices was obtained with ModelFinder (Kalyaanamoorthy et al., 2017) in the program IQTree version 2 (Minh et al., 2020). For both matrices we conducted maximum likelihood (ML) phylogenetic analyses using the same version of the program IQTree with 1,000 ultra-fast bootstrap (BTP) replicates.
We carried out the divergence times estimates for the complete mitogenome matrix with the program BEAST2 version 2.4.7 (Bouckaert et al., 2014) using the Birth-Death model (Nee, 1994) and an uncorrelated lognormal relaxed clock (Drummond et al., 2006). A single partition was considered, using the GTR + I + G model of evolution. To calibrate the tree, we used the mutation rate reported by Papadopoulou et al. (2010) for the mt genome (2.4% per My; 0.012 ± 0.0012). The analysis ran for 100 million generations, sampling trees every 10,000 generations with a burn-in of 25%. The Maximum Clade Credibility Tree (MCCT) was obtained with TreeAnnotator version 1.7.4 (Drummond et al., 2012).
Results
We obtained 7 complete and 9 partial mt genomes with lengths between 13,056 and 15,785 base pairs, and a GC proportion between 25.0 and 26.5% (Table 2). Of the 16 mitogenome sequences, 12 were recovered as a single contig, whereas the remaining 4 were recovered in 2 or more fragments. We did not find arrangement variation in any of the assembled mitogenomes, with all of them being similar to the Pancrustacea ground pattern (Boore et al., 1998) (Fig. 2, Appendix 1).
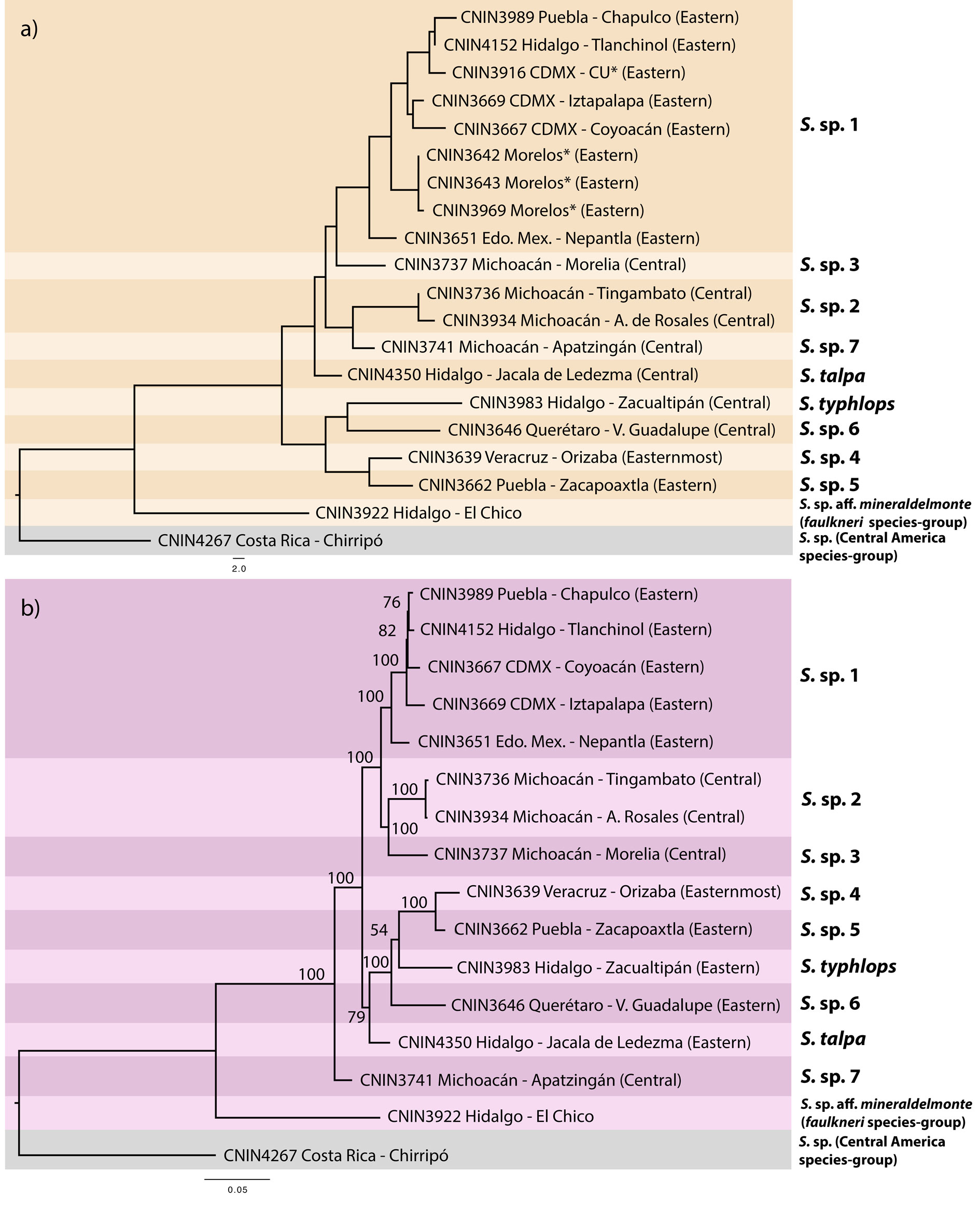
Uncorrected genetic distances for the cox1 and complete mitogenome matrices are shown in Table 3. The pairwise distances observed for the complete mt genome data set generally were slightly lower compared to the ones obtained with cox1. The NJ tree built with the cox1 data set (Fig. 3a) grouped the ingroup taxa in 9 separate clusters that were mostly congruent with the sequence divergence gap of 2%. One of the cases of incongruence was found within the putative S. sp. 1 from the Valley of Mexico, where 6 of the 8 pairwise comparisons that involved the specimen from Nepantla, Estado de Mexico (CNIN3669) were marginally higher than 2% (2.1-2.59%). Furthermore, the only sample from Apatzingán, in the state of Michoacán (S. sp. 7; CNIN3741), had a distance slightly lower than 2% with a sample of S. sp. 1 from Mexico City (CNIN3669; 1.87%) and the sample belonging to S. sp. 3 from Morelia, Michoacán (CNIN3737; 1.9%).
The ML phylograms derived from the complete mt genome and protein-coding gene matrices showed identical relationships, only varying in the BTP support values of some of their clades (Fig. 3b and Appendix 2, respectively). The putative species S. sp. 7 from Apatzingán, Michoacán, in the northernmost part of the Sierra Madre del Sur (SMS) province was sister to the remaining ingroup taxa (BTP = 100), which appeared divided into 2 main clades. One of these clades (BTP = 79) had S. talpa, which is distributed in the limits between the SMO and the TMVB provinces, as sister to the taxa from the eastern part of the TMVB. The second main clade (BTP = 100) contained the putative species from the central portion of the TMVB in the Valley of Mexico and central Michoacán.
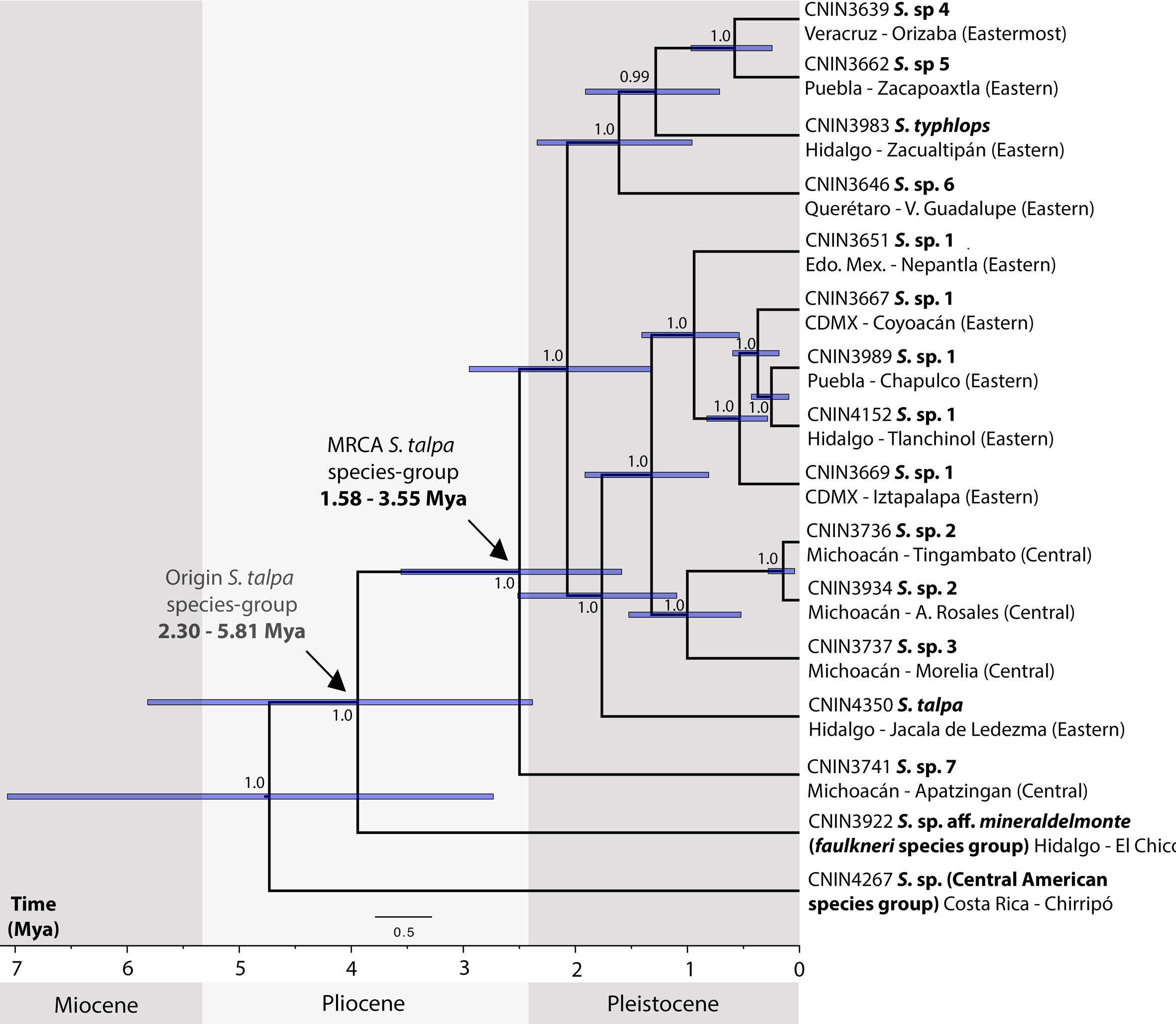
The reconstructed chronogram (Fig. 4) shows that the origin of the S. talpa species-group took place during the late Miocene to late Pliocene, 5.81-2.38 Mya, whereas the divergence between the putative undescribed species from the northernmost portion of the SMS in Apatzingán, Michoacán, and those from the TMVB and SMO provinces probably occurred during the late Pliocene 3.55-2.5 Mya. The split of the members from the central and eastern portions of the TMVB on the other hand probably occurred during the late Pliocene to early Pleistocene, between 2.94 and 1.32 Mya.
Discussion
Mitogenome features in the talpa species-group
Although the Orthoptera is one of the orders whose species are known to have among the largest nuclear insect genomes and thus are more prone to the insertion of nuclear mitochondrial paralogs (Song et al., 2014), its mt gene arrangement has remained generally stable through time (Cameron, 2014; Song et al., 2015). Currently, the only mt gene rearrangement that has been reported as a probable synapomorphy at the supraspecific level within the order is the trnD–trnK tRNA gene translocation within Caelifera (Song et al., 2015).
Here, we have generated and assembled the complete and nearly complete mt genome of various Stenopelmatus specimens, a poorly-known group of ensiferan orthopterans mainly distributed across the Mesoamerican highlands. Previous to this study, only one mitogenome had been published for the genus and the whole family Stenopelmatidae, i.e., Ammopelmatus fuscus Haldeman, 1852 (Song et al., 2015). Thus, our results provide valuable information for a better understanding of the molecular evolution of this orthopteran family and Ensifera in general.
Table 3
Uncorrected distances for the complete mt genome (above) and cox1 (below) data sets that were obtained for the putative species belonging to the S. talpa species-group. Bold numbers are cases of incongruence based on the 2% pairwise distance criterion employed for the cox1 barcoding locus (see results).
| Species | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| 1. S. sp. 1 (Valley of Mexico) | 0-2.59 | 3.85-4.43 | 3.73-4.29 | 5.47-6.08 | 5.3-5.77 | 4-57-4.99 | 5.29-5.82 | 2.47-3.18 | 2.76-3.5 | 9.61-10.24 | 11.88-12.81 |
| 2. S. sp. 2 (Tingambato, Ario de Rosales, Michoacán) | 3.8-5.01 | 0.45 | 3.98-4.08 | 5.82-6.45 | 5.62-5.64 | 5.13-5.32 | 5.97-6.05 | 2.97-3.03 | 3.57-3.61 | 10.31-10.47 | 12.38-12.88 |
| 3. S. sp. 3 (Morelia, Michoacán) | 2.6-3.95 | 3.95-4.4 | – | 6.36 | 5.83 | 4.91 | 6.18 | 2.75 | 3.52 | 10.26 | 12.6 |
| 4. S. sp. 4 (Orizaba, Veracruz) | 5.17-6.23 | 5.77 | 5.77 | – | 2.28 | 4.91 | 5.62 | 4.51 | 4.23 | 10.53 | 12.71 |
| 5. S. sp. 5 (Zacapoaxtla, Puebla) | 5.21-6.53 | 5.62-6.07 | 6.07 | 2.12 | – | 4.16 | 4.91 | 3.77 | 3.74 | 10.18 | 12.55 |
| 6. S. sp. 6 (Valle de Guadalupe, Querétaro) | 5.24-6.29 | 6.43-6.59 | 6.14 | 5.36 | 5.36 | – | 4.59 | 3 | 3.29 | 8.92 | 11.32 |
| 7. S. typhlops (Zacualtipán, Hidalgo) | 5.68-6.83 | 6.68-6.83 | 6.99 | 5.77 | 5.77 | 5.84 | – | 4.15 | 4.16 | 10.16 | 12.62 |
| 8. S. sp. 7 (Apatzingán, Michoacán) | 1.9-3.13 | 2.51-2.83 | 1.87 | 4.72 | 4.85 | 5.54 | 5.65 | – | 19.6 | 7.13 | 10 |
| 9. S. talpa (Jacala, Hidalgo) | 2.33-3.39 | 3.1-3.25 | 2.78 | 4.31 | 4.16 | 4.34 | 4.15 | 2.04 | – | 8.52 | 10.98 |
| 10. S. sp. aff. mineraldelmonte (faulkneri species-group) | 10.05-11.56 | 10.65 | 9.87 | 10.94 | 11.26 | 11.53 | 10.96 | 7.67 | 9.2 | – | 9.97 |
| 11. S. sp. (Central American species-group) | 11.27-12.61 | 11.44-12.31 | 12.15 | 11.85 | 12.31 | 11.79 | 11.85 | 9.54 | 10.6 | 11.09 | – |
Our results indicate that all the assembled mt genomes follow the ancestral insect mt genome structure (Boore et al., 1998), thus supporting the high conservativeness of the mt genome structural organization within Orthoptera. Also, other mt genome features among the examined species, such as the conservation of the trnK-trnD pattern of the tRNAs between the protein-coding genes cox2 and atp8, appear to be similar to those described for other ensiferans (Fenn et al., 2008; Song et al., 2015) as well as a high AT content reported for orthopterans in general (Fen et al., 2008; Sheffield et al., 2010; Song et al., 2015). Further assembling of additional mt genomes of other stenopelmatid taxa will reveal whether the ancestral mt gene organization and other relevant mt features persist in all members of the family.
Barcoding-based species delimitation
A recent molecular phylogenetic study based on mt and 3RAD nuclear data showed the existence of an extensive, highly neglected species diversity in Stenopelmatus (Gutiérrez-Rodríguez et al., 2022). Of the 34 species that were delimited in the latter study, 8 belonged to a clade named the S. talpa species-group, whose species are characterized by having a large size and a generally reddish-orange and black color pattern (although it appears to be exclusively black in S. typhlops; Gutiérrez-Rodríguez et al., 2022).
Our inclusion of additional specimens from the same and newly sampled localities with respect to the above study resulted in 9 barcoding-based species, although the delimitation of samples from Mexico City and surrounding localities decreased to only one putative species in comparison with Gutierrez-Rodríguez et al.’s (2022) study, which discriminated 3 potential evolutionary lineages. The species delineation obtained here, however, must be taken with caution, since some of the cox1 distances that involved specimens from the populations that had been regarded as a different lineage (Nepantla, Estado de Mexico) in some cases exceeded the 2% barcoding gap. Moreover, the specimen from Orizaba, Veracruz was also shown to represent a separate species. This strongly suggests the validity of S. mexicanus Saussure, 1859, a name that was recently regarded as nomen dubium and whose lectotype female was collected in Córdoba, which is less than 25 km in a straight line from Orizaba. A further molecular and morphological examination for all the aforementioned populations will help to consistently establish their species boundaries and taxon assignation.
The relationships obtained here for the members of the S. talpa species-group based on mt genome DNA sequence data were mainly similar to those recovered in a recent phylogenetic study of Stenopelmatus based on a fragment of the cox1 mtDNA gene and nuclear 3RAD data (Gutierrez-Rodríguez et al., 2022). Our divergence times estimates, however, were considerably younger than those obtained by Gutierrez-Rodríguez et al. (2022), probably due to the different source of calibration that they employed. We calibrated our molecular clock analysis taking advantage of a widely employed mutation rate that has been established for the mt genome (2.4% per My; Papadopoulou et al., 2010). In contrast, the above study employed a secondary calibration for the cox1 dataset for the time of divergence between Stenopelmatidae and Anostostomatidae Saussure, 1859 (117.4 My; Song et al., 2015) instead of using a mutation rate, since the use of single mitochondrial markers for deep phylogenies is prone to have considerably high levels of nucleotide saturation (Molak & Ho, 2015). Below we thus make some inferences about the biogeography of the S. talpa species-group based on the relationships and the times of divergence obtained here, which support its recent origin and species diversification (Weissman et al., 2021).
The TMVB is a 1,000 km long Neogene continental arc that is situated across central Mexico, which became an independent province from other partially overlapping arcs during the early to middle Miocene ~10 Mya, originating from the eastward subduction of the Farallon plate beneath western Mexico (Ferrari et al., 2012). The TMVB is characterized by having an east-west (E-W) orientation, being divided into 4 distinct episodes: 1) early- to mid-Miocene, 2) late Miocene, 3) late Miocene-early Pliocene, and 4) late Pliocene and Pleistocene (Ferrari et al., 2012; Gómez-Tuena et al., 2007). In addition, this province is divided into 4 sectors according to their age of inception: Easternmost (~16 Mya), Eastern (~19 Ma), Central (~11 Ma) and Western (~11 Ma) (Ferrari et al., 2012).
According to our results, the S. talpa species-group apparently followed an east-central diversification pattern along the TMVB province. The earliest divergence within the S. talpa species-group was estimated to occur ca. 3.55-1.58 Mya and involved S. sp. 7, a species distributed between the SMS and TMVB border, followed by the clade with the species from the TMVB and adjacent areas of the SMO provinces, which started to diverge ca. 2.94-1.32 Mya. Moreover, most of the earlier divergences within the group occurred along the northern borders of the Eastern sector, while the most recent ones can be found across the Eastern and Central sectors. This is congruent with the aforementioned topographic evolution of the TMVB, which began in the early to mid-Miocene and finished in the late Pliocene to Pleistocene.
The biogeographic inferences drawn here for this apparently recently diverged group of orthopterans endemic to central Mexico will serve as a basis for future evolutionary studies of other groups of organisms with a similar geographic distribution. This work also highlights the utility that the mt genome sequence data have for phylogenetic reconstruction studies of animal taxa, and advocates for more studies that make use of this valuable, highly informative locus.
Acknowledgments
We thank M. García París, N. Percino, K. Guevara, E. K. López-Estrada, L. Escalante Jiménez, J. Ponce-Saavedra, G. Beltrán, M. Rosas-Anaya, C. Pedraza, E. Recuero, and Cristina Mayorga for donating specimens and assistance during fieldtrips; Cristina Mayorga and Guillermina Ortega for their help in the curation of the specimens at CNIN IB-UNAM; and Laura Márquez, Nelly López, and Andrea Jiménez for their help in the laboratory. This study was funded by grants given by DGAPA-UNAM (PAPIIT convocatoria 2019; project Núm. IN201119) by the Consejo Nacional de Ciencia y Tecnología (Conacyt, Mexico, convocatoria Ciencia de Forntera 2019, proyecto Núm. 58548), and by the Dirección General de Cómputo y de Tecnologías de Información y Comunicación, Universidad Nacional Autónoma de México (DGTIC, UNAM; LANCADUNAM-DGTIC-339) to AZR. PXRM was supported by a scholarship given by
the DGTIC, UNAM.
References
Avise, J. C., Arnold, J., Ball, R. M., Bermingham, E., Lamb, T., Neigel, J. E. et al. (1987). Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annual Review of Ecology and Systematics, 18, 489–522.
Bae, J. S., Kim, I., Sohn, H. D., & Jin, B. R. (2004). The mitochondrial genome of the firefly, Pyrocoelia rufa: complete DNA sequence, genome organization, and phylogenetic analysis with other insects. Molecular Phylogenetics and Evolution, 32, 978–985. https://doi.org/10.1016/j.ympev.2004.03.009
Ballard, J. W. O., & Pichaud, N. (2014). Mitochondrial DNA: more than an evolutionary bystander. Functional Ecology, 28, 218–231. https://doi.org/10.1111/1365-2435.12177
Ballard, J. W. O., & Rand, D. M. (2005). The population biology of mitochondrial DNA and its phylogenetic implications. Annual Review of Ecology, Evolution, and Systematics, 36, 621–642. https://doi.org/10.1146/annurev.ecolsys.36.091704.175513
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G. et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution, 69, 313–319. https://doi.org/10.1016/j.ympev.2012.08.023
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Research, 27, 1767–1780. https://doi.org/10.1093/nar/27.8.1767
Boore, J. L., Collins, T. M., Stanton, D., Daehler, L. L., & Brown, W. M. (1995). Deducing the pattern of arthropod phylogeny from mitochondrial DNA rearrangements. Nature, 376, 163–165. https://doi.org/10.1038/376163a0
Boore, J. L., Lavrov, D. V., & Brown, W. M. (1998). Gene translocation links insects and crustaceans. Nature, 392, 667–668. https://doi.org/10.1038/33577
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C. H., Xie, D. et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. Plos Computational Biology, 10, e1003537. https://doi.org/10.1371/journal.pcbi.1003537
Cameron, S. L. (2014). Insect mitochondrial genomics: implications for evolution and phylogeny. Annual Review of Entomology, 59, 95–117. https://doi.org/10.1146/annurev-ento-011613-162007
Cigliano, M. M., Braun, H., Eades, D. C., & Otte, D. (2022). Orthoptera Species File. Version 5.0/5.0. [6th of November, 2022]. http://Orthoptera.SpeciesFile.org
DeSalle, R., Schierwater, B., & Hadrys, H. (2017). MtDNA: The small workhorse of evolutionary studies. Frontiers in Bioscience, 22, 873–887. https://doi.org/10.2741/4522
Drummond, A. J., Ho, S. Y. W., Phillips, M. J., & Rambaut, A. (2006). Relaxed phylogenetics and dating with confidence. Plos Biology, 4, e88. https://doi.org/10.1371/journal.pbio.
0040088
Drummond, A. J., Suchard, M. A., Xie, D., & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973. https://doi.org/10.1093/molbev/mss075
Fenn, J. D., Song, H., Cameron, S. L., & Whiting, M. F. (2008). A preliminary mitochondrial genome phylogeny of Orthoptera (Insecta) and approaches to maximizing phylogenetic signal found within mitochondrial genome data. Molecular Phylogenetics and Evolution, 49, 59–68. https://doi.org/10.1016/j.ympev.2008.07.004
Ferrari, L., Orozco-Esquivel, T., Manea, V., & Manea, M. (2012). The dynamic history of the Trans-Mexican Volcanic Belt and the Mexico subduction zone. Tectonophysics, 522, 122–149. https://doi.org/10.1016/j.tecto.2011.09.018
Glenn, T. C., Nilsen, R. A., Kieran, T. J., Finger, J. W., Pierson, T. W., Bentley, K. E. et al. (2016). Adapterama I: universal stubs and primers for thousands of dual-indexed Illumina libraries (iTru & iNext). PeerJ, 7, e775. https://doi.org/10.7717/peerj.7755
Gómez-Tuena, A., Orozco-Esquivel, M. T., & Ferrari, L. (2007). Igneous petrogenesis of the Trans-Mexican volcanic belt. Geological Society of America Special Papers, 422, 129–181. https://doi.org/10.18268/bsgm2005v57n3a2
Gorochov, A. V. (2021). The Families Stenopelmatidae and Anostostomatidae (Orthoptera). 1. Higher Classification, new and little known taxa. Entomological Review, 100, 1106–1151. https://doi.org/10.1134/S0013873820080084
Gutiérrez-Rodríguez, J., Zaldívar-Riverón, A., Weissman, D. B., & Vandergast, A. G. (2022). Extensive species diversification and marked geographic phylogenetic structure in the Mesoamerican genus Stenopelmatus (Orthoptera: Stenopelmatidae: Stenopelmatinae) revealed by mitochondrial and nuclear 3RAD data. Invertebrate Systematics, 36, 1–21. https://doi.org/10.1071/IS21022
Hajibabaei, M., Singer, G. A., Hebert, P. D., & Hickey, D. A. (2007). DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends in Genetics, 23, 167–172. https://doi.org/10.1016/j.tig.2007.
02.001
Hebert, P. D., Ratnasingham, S., & De Waard, J. R. (2003). Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proceedings of the Royal Society of London Series B: Biological Sciences, 270, S96–S99. https://doi.org/10.1098/rsbl.2003.0025
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., Yi, T. S., & Li, D. Z. (2018). GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. Genome Biology, 21, 1–31. https://doi.org/10.1186/s13059-020-02154-5
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., Von Haeseler, A., & Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods, 14, 587–589. https://doi.org/10.1038/nmeth.4285
Katoh, K., & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. https://doi.org/10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S. et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Lumley, L. M., & Sperling, F. A. (2010). Integrating morpho-
logy and mitochondrial DNA for species delimitation within the spruce budworm (Choristoneura fumiferana) cryptic species complex (Lepidoptera: Tortricidae). Systematic Entomology, 35, 416–428. https://doi.org/10.
1111/j.1365-3113.2009.00514.x
Maddock, S. T., Briscoe, A. G., Wilkinson, M., Waeschenbach, A., San Mauro, D., Day, J. J. et al. (2016). Next-generation mitogenomics: a comparison of approaches applied to caecilian amphibian phylogeny. Plos One, 11, e0156757. https://doi.org/10.1371/journal.pone.0156757
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A. et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37, 1530–1534. https://doi.org/10.1093/molbev/msaa015
Molak, M., & Ho, S. Y. (2015). Prolonged decay of molecular rate estimates for metazoan mitochondrial DNA. PeerJ, 3, e821. https://doi.org/10.7717/peerj.821
Nee, S., May, R. M., & Harvey, P. H. (1994). The reconstructed evolutionary process. Philosophical Transactions of the Royal Society of London Series B: Biological Sciences, 344, 305–311. https://doi.org/10.1098/rstb.1994.0068
Papadopoulou, A., Anastasiou, I., & Vogler, A. P. (2010). Revisiting the insect mitochondrial molecular clock: the mid-Aegean trench calibration. Molecular Biology and Evolution, 27, 1659–1672. https://doi.org/10.1093/molbev/msq051
Saitou, N., & Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425. https://doi.org/10.1093/oxfordjournals.molbev.a040454
Sheffield, N. C., Hiatt, K. D., Valentine, M. C., Song, H., & Whiting, M. F. (2010). Mitochondrial genomics in Orthoptera using MOSAS. Mitochondrial DNA, 21, 87–104. https://doi.org/10.3109/19401736.2010.500812
Song, H., Amédégnato, C., Cigliano, M. M., Desutter-Gandcolas, L., Heads, S. W., Huang, Y. et al. (2015). 300 million years of diversification: elucidating the patterns of orthopteran evolution based on comprehensive taxon and gene sampling. Cladistics, 31, 621–651. https://doi.org/10.1111/cla.12116
Song, H., Moulton, M. J., & Whiting, M. F. (2014). Rampant nuclear insertion of mtDNA across diverse lineages within Orthoptera (Insecta). Plos One, 9, e110508. https://doi.org/10.1371/journal.pone.0110508
Swofford, D. L. (2002). PAUP*. Phylogenetic Analysis Using Parsimony (*and other Methods). Version 4. Sinauer Associates, Sunderland, Mass.
Vandergast, A. G., Weissman, D. B., Wood, D. A., Rentz, D. C., Bazelet, C. S., & Ueshima, N. (2017). Tackling an intractable problem: Can greater taxon sampling help resolve relationships within the Stenopelmatoidea (Orthoptera: Ensifera)? Zootaxa, 4291, 1–33. https://doi.org/10.11646/zootaxa.4291.1.1
Weissman, D. B. (2001). North and Central American Jerusalem crickets (Orthoptera: Stenopelmatidae): taxonomy, distribution, life cycle, ecology and related biology of the American species. In H. Laurence (Ed.), The biology of wetas, king crickets and their allies (pp. 57–72). New York: CAB International. https://doi.org/10.1079/9780851994086.0057
Weissman, D. B. (2005). Jerusalem! cricket? (Orthoptera: Stenopelmatidae: Stenopelmatus); origins of a common name. American Entomology, 51, 138–139. https://doi.org/
10.1093/ae/51.3.138
Weissman, D. B., Vandergast, A. G., Song, H., Shin, S., McKenna, D. D., & Ueshima, N. (2021). Generic relationships of New World Jerusalem crickets (Orthoptera: Stenopelmatoidea: Stenopelmatinae), including all known species of Stenopelmatus. Zootaxa, 4917, 1–122. https://doi.org/10.11646/zootaxa.4917.1.1
