
Jesús Damián Cordero-Ramírez a, Alejandro Miguel Figueroa-López b, Juan Carlos Martínez-Álvarez c, Melina López-Meyer c, Claudia Castro-Martínez c, Juan José Morales-Aguilar d, Ignacio Eduardo Maldonado-Mendoza c, *
a Universidad Autónoma de Occidente, Unidad Regional Guasave, Departamento de Ciencias Naturales y Exactas, Avenida Universidad s/n Colonia Villa Universidad, 81048 Guasave, Sinaloa, Mexico
b Instituto Tecnológico de Sonora, Departamento de Biotecnología y Ciencias Alimentarias, 5 de Febrero 818 Sur, 85000 Ciudad Obregón, Sonora, Mexico
c Instituto Politécnico Nacional, Centro Interdisciplinario de Investigación para el Desarrollo Integral Regional – Unidad Sinaloa, Departamento de Biotecnología Agrícola, Laboratorio de Ecología Molecular de la Rizósfera, Boulevard Juan de Dios Bátiz Paredes Núm. 250, Col. San Joachin, 81101 Guasave, Sinaloa, Mexico
d Universidad Autónoma de Occidente, Unidad Regional Guasave, Departamento de Ciencias de la Salud, Avenida Universidad s/n Colonia Villa Universidad, 81048 Guasave, Sinaloa, Mexico
*Corresponding author: imaldona@ipn.mx (I.E. Maldonado-Mendoza)
Received: 11 June 2020; accepted: 26 September 2021
Abstract
Rhizospheric microbiota diversity of crops in agroecosystems is understudied in Mexico and worldwide. The aim of the present work was to explore the diversity of culturable bacteria in maize fields. A bacterial collection consisting of 11,520 purified isolates was created from the rhizosphere of maize plants. Genomic DNA was obtained from each isolate and a region of 16S rDNA was sequenced. The 16S rDNA amplicon sequences were analyzed and grouped into Operational Taxonomic Units (OTUs), allowing the assemblage of 7,077 bacterial isolates into 185 non-singleton OTUs. OTUs belonged to 19 bacterial genera within Firmicutes, Proteobacteria, Actinobacteria and Bacteroidetes Phyla; with Firmicutes as the richest Phylum comprising 146 OTUs and 6 genera, and being Bacillus the richest genus. The soil core-community of 28 OTUs belonging to Firmicutes and 1 OTU from Proteobacteria was identified. The work discusses the role that the different bacterial populations identified within the maize rhizosphere may play, their potential use for biotechnological purposes, and the importance of conservation of microbiological resources using bacterial collections.
Keywords: Culturable microbiota; Maize; Bacterial populations; Microbial collections
© 2022 Universidad Nacional Autónoma de México, Instituto de Biología. This is an open access article under the CC BY-NC-ND license
Bacterias cultivables de la rizósfera del maíz:
conservando el potencial de los recursos biotecnológicos mexicanos
Resumen
La diversidad de la microbiota asociada a la rizósfera de cultivos en sistemas agrícolas ha sido pobremente estudiada en México y en todo el mundo. El objetivo de este trabajo fue identificar la diversidad de bacterias cultivables en campos de maíz. Se creó una colección de cepas de 11,520 aislados purificados a partir de la rizósfera de maíz. Se procedió a extraer el ADN genómico y secuenciar una región del 16S rADN de cada aislado. Las secuencias fueron analizadas y agrupadas en unidades operacionales taxonómicas (OTU). Esto permitió la agrupación de 7,077 cepas en 185 OTU pertenecientes a 19 géneros dentro de los phyla Firmicutes, Proteobacteria, Actinobacteria y Bacteroidetes; siendo Firmicutes el phylum más rico, incluyendo 146 OTU y 6 géneros, con Bacillus como el género con más especies. Se identificó la comunidad núcleo de los suelos conteniendo 28 OTU de Firmicutes y 1 OTU de Proteobacteria. Se discute el papel que juegan las diferentes poblaciones bacterianas identificadas en la rizósfera del maíz, su potencial para ser empleadas con propósitos biotecnológicos y la importancia de la conservación de los recursos microbianos empleando colecciones bacterianas.
Palabras clave: Microbiota cultivable; Maíz; Poblaciones bacterianas; Colecciones microbianas
© 2022 Universidad Nacional Autónoma de México, Instituto de Biología. Este es un artículo Open Access bajo la licencia CC BY-NC-ND
Introduction
Plant roots provide a nutrient-rich environment for a large number of soil microorganisms. The rhizosphere, the zone in close proximity to the root surface, typically contains 10 to 100 times more microorganisms per gram than bulk soil (Haas et al., 2002). The organisms harbored by the rhizosphere can have a neutral, deleterious or beneficial effect on the plant. Different factors, such as land use, soil type, soil texture, pH, nitrogen availability and plant species affect the bacterial community structure (reviewed in Saleem et al., 2019). Furthermore, diversity and stability of plant-associated bacterial communities influence soil and plant quality, as well as ecosystem sustainability. Several studies have examined the effect of phytopathogens on the microbial diversity of plant-associated bacteria (Li et al., 2014; Mendes et al., 2011). These reports show that the presence of plant pathogens influences population dynamics in the rhizosphere, and that in some cases certain bacterial groups might affect disease development. Therefore, the analysis of the plant microbiota will allow identifying microorganisms with potential for plant growth promotion, phytopathogen control and bioactive compounds production for potential biotechnological applications (Douriet-Gámez et al., 2018; Figueroa-López et al., 2016; Ibarra-Galeana et al., 2017).
In Sinaloa, Mexico, particularly in Guasave Valley, natural vegetation is scarce, since most of the soil surface (~ 200,000 ha) is used for agriculture. There are very small conserved zones that show relictual vegetation of low deciduous forest (H. Ayuntamiento de Guasave, 1998). Agriculture in Sinaloa is highly technified. This is the highest maize producing state with around 0.5 million ha cultivated yearly under the scheme of intensive monoculture producing around 6 million metric tons of grain per year (SIAP, 2020). Soils submitted to monoculture for many years are highly susceptible to the loss of biological diversity (Liu et al., 2020; Trejo-Aguilar et al., 2013). Maize is affected by different biotic stresses including diverse bacterial and fungal pathogens that are currently treated with different agrochemicals and could be treated using biological control. This is the case of Fusarium verticillioides, a fungal pathogen causing stalk and ear rot of maize and being controlled by Bacillus cereus B25 (Lizárraga-Sánchez et al., 2015). A more complete understanding of the microbial ecology and diversity associated with the maize rhizosphere could help to improve plant health in field crops, reduce our dependence on chemical pesticides used in agriculture, and develop efficient biological control strategies. Therefore, it is important to characterize the microbial communities naturally associated with maize root systems, to identify potential growth promotion agents. To address this, studies have focused on characterizing rhizospheric maize bacterial communities using non-cultivation approaches and employing new sequencing technologies (Pereira et al., 2011). However, there is a drawback to metagenomic studies performed on DNA soil samples: dormancy allows bacteria to persist during unfavorable conditions, and surveys estimate that over 80% of the bacterial cells in the soil are dormant (Lennon & Jones, 2011). Moreover, the community of physiologically active bacteria within the soil is distinct from those that are dormant (Lennon & Jones, 2011). As a result, estimates of bacterial composition using standard DNA extractions from soil may not provide measurements that reflect the active players in the plant-microbe interaction, potentially obscuring field attempts to identify the agents of microbial control (Bulgarelli et al., 2013).
In this work, special consideration was given to the discovery of bacterial isolates from corn fields in Sinaloa. These native isolates are already pre-adapted to the prevailing edaphic and climatological conditions in this region. Additionally, co-existence for many years with the natural soil microbiota should provide these microorganisms with competitive advantages compared to recently introduced exotic species. This collection has been previously utilized for identifying native bacteria capable of exerting biocontrol against the fungal phytopathogen F. verticillioides (Figueroa-Lopez et al., 2014, 2016). Bacteria with the highest biocontrol potential have been tested in field trials with great success (Lizárraga-Sánchez et al., 2015). As a result of this work, we have identified Bacillus cereus sensu lato strain B25 as the best F. verticillioides antagonist. B25 displays a whole array of antagonistic mechanisms that can be used to control F. verticillioides growth (Douriet-Gámez et al., 2018), including B25 chitinases which expression is induced in response to F. verticillioides lysates and might be involved in direct control of F. verticillioides conidia germination (Figueroa-López et al., 2017; Morales-Ruiz et al., 2021). In addition, we have characterized in this collection phosphate-solubilizing bacteria with the potential for growth and phosphorus nutrition improvement in maize. We found 2 Bacillus species: B. flexus (B4) and B. megaterium (B5) as potential phosphate solubilizers associated to the maize rhizosphere (Ibarra-Galeana et al., 2017). This collection has been kept under scrutiny to find diverse isolates for different biotechnological purposes. For these reasons and because biotechnological use requires live organisms, our aim was to obtain culturable bacteria from the maize rhizosphere to examine the diversity of culturable bacterial populations. These will help us to identify bacterial strains with biotechnological potential and preserve them for further use.
Materials and methods
We sampled 5 locations in northern Sinaloa, Mexico, differing by planting date and by maize hybrid: 1) Serrano (Salvador Alvarado municipality), 2) Alhuey (Guasave municipality), 3) 18 de Diciembre (Angostura municipality), 4) Casa Blanca (Guasave municipality), and 5) La Trinidad (Guasave municipality). In each location, 10 maize rhizosphere and bulk-soil samples were collected corresponding to 10 plants. Three to 4 kg of bulk soil were removed with a shovel from the stem base of each plant (0-30 cm in depth). The soil samples were transferred to the laboratory at room temperature and processed immediately. Soil particles adhering to the roots (rhizospheric soil) were collected. In both cases, rhizospheric and bulk soil samples from the 10 plants from each location were mixed thoroughly by using sub-samples of 1 kg for bulk-soil and 10 g for rhizospheric samples to prepare a composite sample by mixing thoroughly the 10 different samples corresponding to the 10 plants per location. Composite samples of bulk-soil were stored at room temperature (25 °C) for physicochemical characterization. Composite rhizosphere samples were used immediately to isolate microorganisms by serial dilutions.
In order to obtain bacterial isolates, 4 culture media were prepared in 100 mm-diameter Petri dishes to enrich for certain specific taxonomic groups: 1) Luria Bertani (LB) medium was used to enrich for Bacillus isolates (Cavaglieri et al., 2005); 2) Actinomycetes Isolation Agar (AIA) (Bressan & Fontes-Figueiredo, 2007); 3) King B Agar (KBA) was used for Pseudomonas enrichment (Cavaglieri et al., 2004); and 4) Man, Rogosa and Sharpe (MRS) medium was used for lactic acid bacteria (De Man et al., 1960). Colonies were collected from the serial dilutions plated on LB, KBA and MRS media after 24 hours growth and from AIA medium after 48-72 hours at 25 ºC.
To generate the maize rhizospheric culturable bacterial collection, 576 isolates were “picked” and arranged in 6 96-well plates from each specific culture medium. This yielded 2,304 isolates from each composite rhizospheric sample points; the complete collection therefore contained 11,520 isolates. Isolates were cryopreserved in triplicate at -70 °C in January 2009, using LB containing 15% glycerol (v/v) according to Pasarell and McGinnis (1992). Frozen stocks were made and 2 months later they were thawed and grown at 25 °C and 200 r/min in 2 mL 96-well plates containing 1.5 mL liquid medium, for either 24 hours (LB, KBA and MRS media) or 72 hours (AIA medium). Isolates were considered non-viable if no visible growth after culturing in the proper media was observed after thawing. Plates were centrifuged at 2,000 rpm for 10 min and the bacterial pellets from viable isolates were kept at -70 °C until processing for DNA extraction.
A bulk soil sub-sample of 500 g was used for nutrient and physicochemical soil analyses (Supplementary material: Table S1). Texture was determined based on soil texture classification by particle size distributions (USDA), phosphate was analyzed according to Olsen et al. (1954), and organic matter was analyzed according to Walkley and Black (1934).
From the total 11,520 isolates, 95% (10,944) were viable, 10,080 isolates out of these were regrown and processed for DNA extraction, PCR amplification of 16S rDNA and sequencing. Bacterial DNA was extracted with the DNeasy® Blood & Tissue Kit (Qiagen; CA, USA). The following primers were used to amplify the complete 16S rDNA region (1,400 pb): F2C (5’- AGA GTT TGA TCA TGG CTC -3’) and C (5’- ACG GGC GGT GTG TAC -3’) (Shi et al., 1997). The 25 μL PCR mixture contained 10 ng of DNA template, 1X reaction buffer, 10 pmol of each primer, 10 µM of each deoxynucleoside triphosphate (dNTP), and 1 U of Taq DNA polymerase (Invitrogen; Carlsbad, CA, USA). The PCR conditions included an initial denaturation step at 95 ºC (4 min); 32 cycles of denaturation at 95 ºC (1 min) followed by annealing at 60 ºC (30 sec) and extension at 72 ºC (1.5 min); and a final step at 72 ºC (5 min). PCR reactions were carried out in 96-well plates using a MyCycler thermal cycler (BioRad; CA, USA). Products were visualized by 1% agarose (w/v) gel electrophoresis in 0.5 X Tris-acetate-EDTA (TAE) buffer and stained with ethidium bromide. PCR products were purified with a QIAquick PCR Purification kit (Qiagen; CA, USA) and quantitated using a Nanodrop 2000 UV-Vis spectrophotometer (Thermo Scientific). Approximately 400 ng of PCR product were used for the sequencing. The U1 primer (5’ – CCA GCA GCC GCG GTA ATA CG – 3’) (Lu et al., 2000) internal to the F2C/C amplicon was used for sequencing with an ABI 3730 XL automated sequencer at the National Laboratory of Genomics (LANGEBIO; Irapuato, Mexico). The resulting sequences ranged from 300 to 600 bp and contained a hypervariable region (domains V1-V3) (Sun et al., 2013).
Isolates were identified by sequence comparison against the GenBank databases and their genera identity were confirmed in the Ribosomal Database Project (RDP) (Cole et al., 2014). Ten thousand and eighty isolates out of 10,944 viable isolates were sequenced. Short (< 200 nt in length) and low-quality sequences were eliminated from the analyses. The resulting high-quality sequences were examined for the presence of chimeric sequences with the CHIMERA_CHECK software, available from the website (http://rdp.cme.msu.edu/); chimeric sequences were then discarded (yielding 7,077 sequences for the next analyses). A total of 7,077 sequences were grouped into operational taxonomic units (OTUs) (Supplementary material: Fig. S1) with the Cluster tool using the average neighbor algorithm from the Mothur program, v. 1.20.1 (http://www.mothur.org) using 0.03, 0.05 differences to construct rarefaction curves (Schloss et al., 2009). All singletons were removed to avoid any bias from minimally represented sequences. A total of 6,569 non-singleton sequences were grouped into OTUs defined by a 97% pairwise similarity threshold.
Sequences were aligned with the Clustal-W program (Thompson et al., 1994). A phylogenetic tree of OTUs representing all genera identified was constructed based on MEGA X (bootstrap = 1000), using the maximum likelihood method, and Tamura-Nei model.
Results
The bacterial strain collection, comprising 11,520 isolates, exhibited 95% survival efficiency 2 months after freezing and thawing, yielding 10,944 isolates. High quality 16S rDNA gene sequences were obtained from 10,080 isolates and after eliminating short, chimeric or low-quality sequences a total of 7,077 sequences with an average length ~ 300-600 nt were recovered for analysis and deposited in GenBank (with accession numbers JQ829081 through JQ836199).
A total of 6,569 non-singleton sequences were grouped in 185 OTUs (Table 1). Rarefaction analysis revealed that the species sampling effort curves calculated with 95% and 97% sequence identity have a positive slope with no evidence of approaching saturation (Supplementary material: Fig. S1).
Identification at the genus level using RDP and GenBank databases was successful for most isolates assigning putative taxonomic identities to each OTU in 19 genera (Table 1, Supplementary material: Table S2). These genera were grouped into 4 different bacteria phyla: Firmicutes, Actinobacteria, Bacteroidetes and Proteobacteria (Fig. 1).
Firmicutes and Proteobacteria displayed the greatest number of OTUs. Within Firmicutes, 146 OTUs were grouped into 6 genera, whereas Proteobacteria were represented by 34 OTUs grouped into 10 genera. Firmicutes was primarily represented by Bacillus and 5 other less abundant genera (Table 1). Notably, most of the putative Proteobacteria OTUs were grouped in the γ-Proteobacteria class. This class was represented mainly by the genera Pseudomonas and Enterobacter (15 and 4 OTUs, respectively) in addition to another 6 genera. The α- and β-Proteobacteria classes were solely represented by 1 OTU each (from the genera Massilia and Rhizobium, respectively) (Table 1).
The least diverse groups were the Actinobacteria and Bacteroidetes. In Actinobacteria we identified 31 and 4 sequences, belonging to the genera Arthrobacter (OTUs 114 and 248) and Sinomonas (OTU 174), respectively. In Bacteroidetes, only 6 sequences belonging to the genus Sphingobacterium were identified (OTU 243) (Table 1).
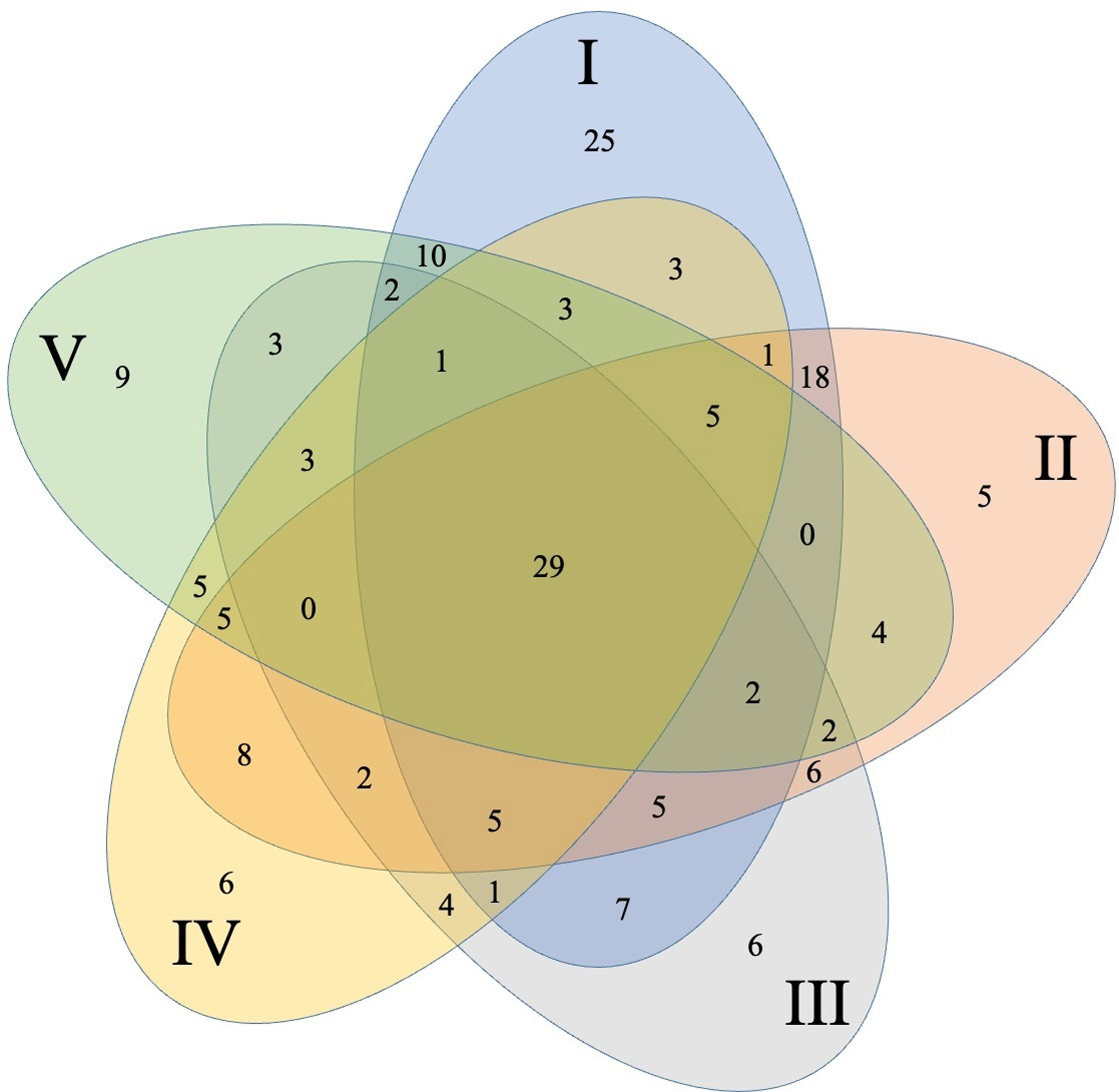
The core community was represented by 29 OTUs (highlighted in Table 1, Fig. 2) which were present in all 5 collection locations, 28 of these belong to Firmicutes with 26 from the genus Bacillus and only 1 OTU from Lysinibacillus and 1 from Paenibacillus. OTU 6 belonging to Bacillus was by far the most abundant one representing 23.69% of the total sequences. Only 1 OTU (36) belonged to γ-proteobacteria within the genus Pseudomonas.
Significant differences were detected between different collection locations in 5 (pH, P, K, Ca, Mg) of the 12-soil chemical or physical parameters evaluated in the bulk-soil samples [Supplementary material: Table S1]). We found significant pH changes between site I and the other sites. Site IV was significantly higher in P and Ca than the other sites. The sampling sites I and V were significantly lower than the remaining 3 points. Sites IV and V were higher on Mg than the remaining 3 points (Supplementary material: Table S1). Within these sites OTUs richness was: site I (117 OTUs) > site II (97) >, site V (83) > site IV (81) > site III (78 OTUs) (Table 1).
Discussion
The goal of our study was to analyze culturable bacterial communities from the maize rhizosphere. Although many purified bacterial isolates were sequenced and analyzed, our sampling effort was insufficient to reach a plateau in a rarefaction analysis of the 16S rDNA sequences; therefore, a complete diversity study of the bacteria present in the maize rhizosphere could not be performed. Among the phyla identified in our study Actinobacteria, Firmicutes and Proteobacteria were predominant; as reported before, the main groups of microorganisms that preferentially colonize maize plants belong to the phyla Actinobacteria, Bacteroidetes, Cyanobacteria, Firmicutes and Proteobacteria (Li et al., 2014; Peiffer et al., 2013). These groups were identified in a microbiome study as the most changing taxa in the rhizospheres of sugar beet (Mendes et al., 2011), maize (Yang et al., 2017) and rice (Zhang et al., 2017) and some species of Pseudomonas (Proteobacteria) are associated with disease suppression of Rhizoctonia solani (Mendes et al., 2011). In agreement with our findings, Firmicutes and Proteobacteria have also been reported as the most abundant and diverse phyla in the maize rhizosphere of 20-day old maize seedlings (Pereira et al., 2011). In these reports, Bacillus and Pseudomonas were the major community components of the maize rhizosphere, using either culture-dependent or independent methods. This is consistent with our findings where Firmicutes was the most abundant bacterial phylum in the maize rhizosphere in this study. Additionally, the core community in this study was integrated mainly by the phylum Firmicutes and in less proportion by the phylum Proteobacteria corresponding well with previous studies that describe the presence of these phyla in the bacterial community of maize rhizosphere (Li et al., 2014; Omotayo et al., 2021; Peiffer et al., 2013). Both, Firmicutes and Proteobacteria have important members with diverse activities such as carbon, sulfur, nitrogen cycling essential for nutrient cycling and are used in phytoremediation, biocontrol of phytopathogens and as plant biofertilizers (Omotayo et al., 2021).
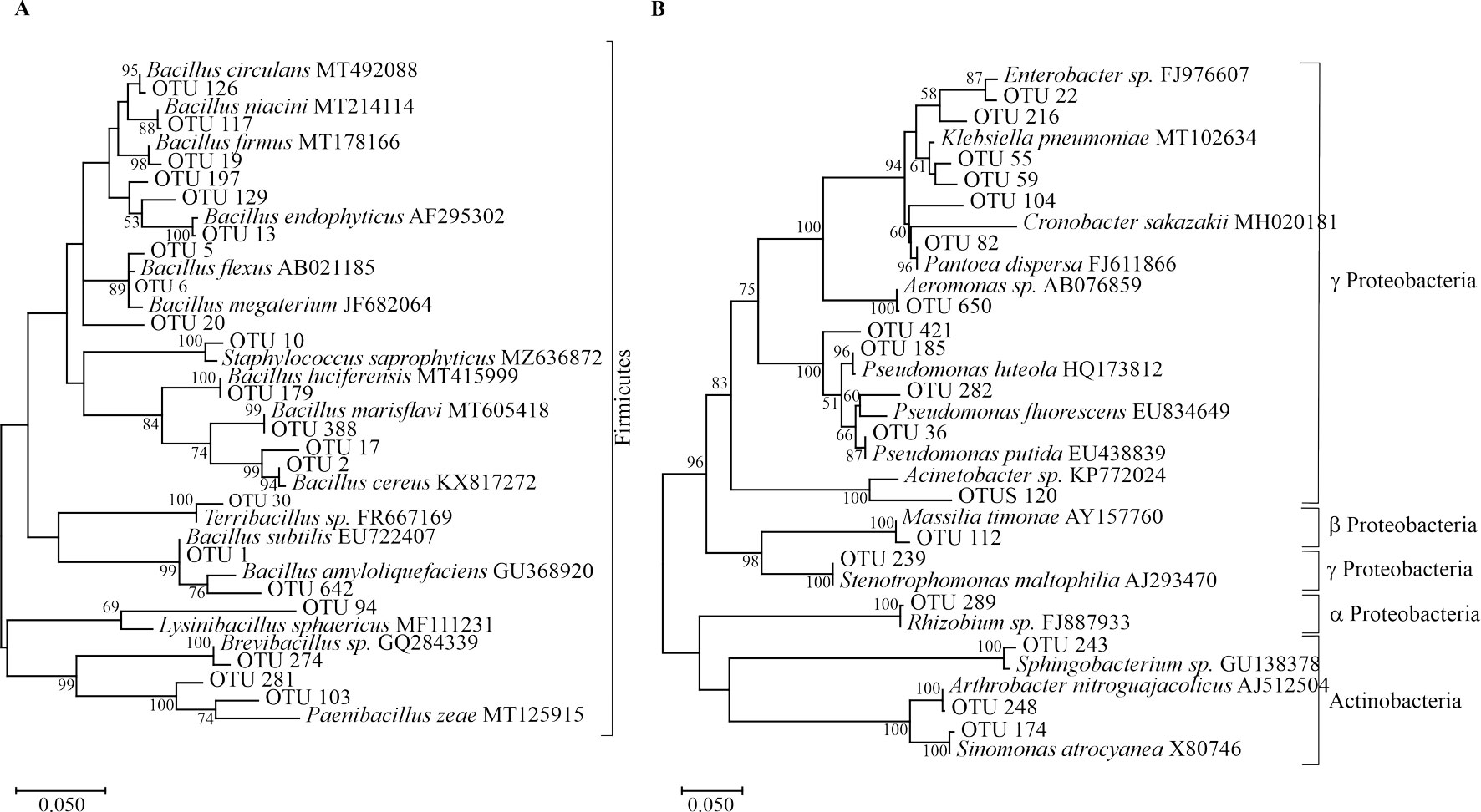
Members of these phyla have been well-described as either plant growth-promoting rhizobacteria (PGPR) or as biocontrol agents (Nagórska et al., 2007). We have previously demonstrated the biotechnological potential of several isolates obtained in the present study. For example, Bacillus cereus sensu lato strain B25 (OTU 2 from this work) increased grain yield following seed bacterization, as compared to a non-inoculated control (Lizárraga-Sánchez et al., 2015). In vitro testing of Bacillus spp. in maize roots and kernels inhibited growth of the phytopathogen Fusarium verticillioides and the production of fumonisin B1, a mycotoxin produced by this fungus (Cavaglieri et al., 2004). B25 has been identified as Bacillus cereus in this study (Fig. 1, OTU 2) and by phylogenomic analysis (Douriet-Gámez et al., 2018). B25 exerts biocontrol of F. verticillioides in vitro, in greenhouse (Figueroa-López et al., 2016) and field experiments. This bacterium also inhibits total fumonisin production in grain (Lizárraga-Sánchez et al., 2015). OTU 2 is highly represented (14.99%) in the culturable bacterial population (985 out of 6,569 16S rDNA sequences) and belongs to the core community of OTUs being present in all sites sampled.
Bacillus megaterium isolated from the maize rhizosphere has been reported to promote growth and development of Phaseolus vulgaris and Arabidopsis thaliana (López-Bucio et al., 2007), as well as B. flexus and B. megaterium (both belonging to OTU 6 from this work) have demonstrated ability for P-solubilization in maize in pot experiments (Ibarra-Galeana et al., 2017). OTU 6 also belongs to the core community of OTUs and is by far the most abundant OTU representing 23.69%, or 1,556 sequences from the culturable bacterial population. Bacillus thuringiensis is used as a biocontrol agent of diverse phytopathogens (Lucon et al., 2010).
Table 1
Maize rhizosphere bacterial OTU distribution and colonies number in five collection sites in Sinaloa Mexico. Data were measured as total number of colonies sequenced per OTU (total sequences) and relative abundance as percentage of the total number of colonies sequenced. Highlighted in gray: OTUs that were present in all soil samples; N/D: not defined.
| Phyla/Class | Genera | OTU | Site I | Site II | Site III | Site IV | Site V | Total sequences | % |
| Actinobacteria | Arthrobacter | OTU 114 | 23 | 3 | 2 | 28 | 0.43 | ||
| Arthrobacter | OTU 248 | 1 | 2 | 3 | 0.05 | ||||
| Sinomonas | OTU 174 | 2 | 1 | 1 | 4 | 0.06 | |||
| Bacteroidetes | Sphingobacterium | OTU 243 | 1 | 5 | 6 | 0.09 | |||
| Firmicutes | Bacillus | OTU 1 | 127 | 200 | 122 | 121 | 39 | 609 | 9.27 |
| Bacillus | OTU 2 | 117 | 172 | 130 | 194 | 372 | 985 | 14.99 | |
| Bacillus | OTU 3 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 4 | 12 | 22 | 13 | 26 | 23 | 96 | 1.46 | |
| Bacillus | OTU 5 | 67 | 33 | 33 | 34 | 24 | 191 | 2.91 | |
| Bacillus | OTU 6 | 425 | 281 | 304 | 370 | 176 | 1,556 | 23.69 | |
| Bacillus | OTU 7 | 17 | 65 | 26 | 43 | 80 | 231 | 3.52 | |
| Bacillus | OTU 8 | 14 | 15 | 7 | 5 | 3 | 44 | 0.67 | |
| Bacillus | OTU 12 | 19 | 35 | 5 | 19 | 8 | 86 | 1.31 | |
| Bacillus | OTU 13 | 2 | 30 | 19 | 3 | 16 | 70 | 1.07 | |
| Bacillus | OTU 15 | 2 | 5 | 3 | 8 | 7 | 25 | 0.38 | |
| Bacillus | OTU 16 | 7 | 4 | 3 | 3 | 3 | 20 | 0.3 | |
| Bacillus | OTU 17 | 9 | 33 | 4 | 18 | 10 | 74 | 1.13 | |
| Bacillus | OTU 18 | 11 | 23 | 2 | 11 | 6 | 53 | 0.81 | |
| Bacillus | OTU 19 | 10 | 25 | 66 | 21 | 30 | 152 | 2.31 | |
| Bacillus | OTU 20 | 1 | 1 | 1 | 3 | 0.05 | |||
| Bacillus | OTU 21 | 79 | 47 | 30 | 48 | 21 | 225 | 3.43 | |
| Bacillus | OTU 23 | 4 | 3 | 1 | 1 | 9 | 0.14 | ||
| Bacillus | OTU 25 | 2 | 5 | 7 | 0.11 | ||||
| Bacillus | OTU 26 | 1 | 3 | 6 | 10 | 0.15 | |||
| Bacillus | OTU 34 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 43 | 4 | 1 | 5 | 2 | 12 | 0.18 | ||
| Bacillus | OTU 51 | 2 | 1 | 3 | 0.05 | ||||
| Bacillus | OTU 52 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 54 | 3 | 3 | 0.05 | |||||
| Bacillus | OTU 60 | 1 | 1 | 1 | 3 | 0.05 | |||
| Bacillus | OTU 79 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 84 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 95 | 65 | 76 | 18 | 53 | 19 | 231 | 3.52 | |
| Bacillus | OTU 98 | 1 | 1 | 2 | 3 | 2 | 9 | 0.14 | |
| Bacillus | OTU 113 | 1 | 3 | 3 | 2 | 10 | 19 | 0.29 | |
| Bacillus | OTU 117 | 41 | 11 | 11 | 15 | 13 | 91 | 1.39 | |
| Bacillus | OTU 118 | 2 | 2 | 4 | 0.06 | ||||
| Bacillus | OTU 121 | 1 | 3 | 4 | 0.06 | ||||
| Bacillus | OTU 123 | 2 | 1 | 3 | 0.05 | ||||
| Bacillus | OTU 124 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 125 | 2 | 3 | 1 | 6 | 0.09 | |||
| Bacillus | OTU 126 | 1 | 3 | 2 | 1 | 1 | 8 | 0.12 | |
| Bacillus | OTU 127 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 128 | 25 | 12 | 14 | 1 | 52 | 0.79 | ||
| Bacillus | OTU 129 | 4 | 4 | 0.06 | |||||
| Bacillus | OTU 130 | 7 | 7 | 5 | 5 | 3 | 27 | 0.41 | |
| Bacillus | OTU 131 | 6 | 3 | 2 | 1 | 4 | 16 | 0.24 | |
| Bacillus | OTU 134 | 4 | 8 | 2 | 5 | 2 | 21 | 0.32 | |
| Bacillus | OTU 137 | 2 | 2 | 1 | 4 | 9 | 0.14 | ||
| Bacillus | OTU 138 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 141 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 151 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 156 | 1 | 1 | 1 | 1 | 4 | 0.06 | ||
| Bacillus | OTU 157 | 2 | 1 | 1 | 4 | 0.06 | |||
| Bacillus | OTU 159 | 2 | 2 | 5 | 5 | 4 | 18 | 0.27 | |
| Bacillus | OTU 162 | 1 | 2 | 3 | 6 | 0.09 | |||
| Bacillus | OTU 163 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 172 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 175 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 179 | 1 | 6 | 1 | 8 | 0.12 | |||
| Bacillus | OTU 183 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 190 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 195 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 196 | 1 | 3 | 4 | 0.06 | ||||
| Bacillus | OTU 197 | 1 | 2 | 3 | 0.05 | ||||
| Bacillus | OTU 202 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 208 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 212 | 1 | 1 | 3 | 5 | 0.08 | |||
| Bacillus | OTU 220 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 224 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 228 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 231 | 1 | 3 | 1 | 5 | 0.08 | |||
| Bacillus | OTU 251 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 252 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 253 | 2 | 1 | 2 | 5 | 0.08 | |||
| Bacillus | OTU 254 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 259 | 1 | 2 | 3 | 0.05 | ||||
| Bacillus | OTU 260 | 2 | 1 | 3 | 0.05 | ||||
| Bacillus | OTU 261 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 262 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 266 | 2 | 2 | 1 | 1 | 1 | 7 | 0.11 | |
| Bacillus | OTU 267 | 11 | 5 | 11 | 8 | 35 | 0.53 | ||
| Bacillus | OTU 268 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 269 | 3 | 1 | 4 | 0.06 | ||||
| Bacillus | OTU 270 | 8 | 3 | 2 | 7 | 3 | 23 | 0.35 | |
| Bacillus | OTU 272 | 2 | 1 | 1 | 4 | 0.06 | |||
| Bacillus | OTU 273 | 1 | 5 | 5 | 3 | 14 | 0.21 | ||
| Bacillus | OTU 275 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 288 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 321 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 324 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 325 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 331 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 336 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 365 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 372 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 381 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 388 | 9 | 17 | 5 | 31 | 0.47 | |||
| Bacillus | OTU 390 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 398 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 401 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 409 | 3 | 3 | 0.05 | |||||
| Bacillus | OTU 411 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 412 | 4 | 4 | 0.06 | |||||
| Bacillus | OTU 416 | 1 | 2 | 3 | 0.05 | ||||
| Bacillus | OTU 423 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 433 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 451 | 3 | 2 | 5 | 0.08 | ||||
| Bacillus | OTU 454 | 1 | 1 | 2 | 4 | 0.06 | |||
| Bacillus | OTU 456 | 2 | 1 | 1 | 4 | 0.06 | |||
| Bacillus | OTU 459 | 2 | 1 | 2 | 1 | 6 | 0.09 | ||
| Bacillus | OTU 464 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 469 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 489 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 490 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 494 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 495 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 505 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 510 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 521 | 1 | 1 | 2 | 1 | 5 | 0.08 | ||
| Bacillus | OTU 523 | 4 | 1 | 5 | 0.08 | ||||
| Bacillus | OTU 541 | 1 | 2 | 3 | 0.05 | ||||
| Bacillus | OTU 567 | 1 | 2 | 1 | 4 | 0.06 | |||
| Bacillus | OTU 568 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 575 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 576 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 577 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 584 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 592 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 593 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 596 | 1 | 1 | 1 | 3 | 0.05 | |||
| Bacillus | OTU 607 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 625 | 1 | 1 | 2 | 0.03 | ||||
| Bacillus | OTU 642 | 2 | 2 | 0.03 | |||||
| Bacillus | OTU 662 | 2 | 2 | 0.03 | |||||
| Brevibacillus | OTU 274 | 1 | 2 | 3 | 0.05 | ||||
| Lysinibacillus | OTU 94 | 29 | 4 | 14 | 56 | 157 | 260 | 3.96 | |
| Lysinibacillus | OTU 375 | 1 | 11 | 12 | 0.18 | ||||
| Lysinibacillus | OTU 385 | 1 | 3 | 2 | 6 | 0.09 | |||
| Lysinibacillus | OTU 392 | 2 | 2 | 0.03 | |||||
| Lysinibacillus | OTU 393 | 2 | 2 | 0.03 | |||||
| Paenibacillus | OTU 103 | 1 | 2 | 5 | 1 | 3 | 12 | 0.18 | |
| Paenibacillus | OTU 281 | 2 | 1 | 1 | 4 | 0.06 | |||
| Paenibacillus | OTU 387 | 3 | 1 | 4 | 0.06 | ||||
| Paenibacillus | OTU 394 | 1 | 3 | 4 | 2 | 10 | 0.15 | ||
| Paenibacillus | OTU 422 | 1 | 1 | 8 | 2 | 12 | 0.18 | ||
| Paenibacillus | OTU 453 | 2 | 2 | 0.03 | |||||
| Staphylococcus | OTU 10 | 1 | 4 | 2 | 1 | 8 | 0.12 | ||
| Terribacillus | OTU 30 | 3 | 6 | 3 | 2 | 14 | 0.21 | ||
| Terribacillus | OTU 441 | 1 | 1 | 2 | 0.03 | ||||
| N/D | OTU 221 | 1 | 1 | 2 | 0.03 | ||||
| α-Proteobacteria | Rhizobium | OTU 289 | 3 | 1 | 4 | 0.06 | |||
| β-Proteobacteria | Massilia | OTU 112 | 2 | 2 | 0.03 | ||||
| γ-Proteobacteria | Acinetobacter | OTU 120 | 1 | 5 | 8 | 14 | 0.21 | ||
| Acinetobacter | OTU 164 | 2 | 2 | 0.03 | |||||
| Acinetobacter | OTU 167 | 2 | 2 | 0.03 | |||||
| Acinetobacter | OTU 169 | 15 | 2 | 17 | 0.26 | ||||
| Aeromonas | OTU 650 | 18 | 18 | 0.27 | |||||
| Cronobacter | OTU 104 | 2 | 2 | 0.03 | |||||
| Enterobacter | OTU 22 | 340 | 20 | 39 | 399 | 6.07 | |||
| Enterobacter | OTU 216 | 2 | 1 | 3 | 0.05 | ||||
| Enterobacter | OTU 335 | 2 | 2 | 0.03 | |||||
| Enterobacter | OTU 368 | 3 | 3 | 0.05 | |||||
| Klebsiella | OTU 55 | 4 | 4 | 0.06 | |||||
| Klebsiella | OTU 59 | 2 | 2 | 0.03 | |||||
| Klebsiella | OTU 313 | 4 | 4 | 0.06 | |||||
| Pantoea | OTU 82 | 68 | 1 | 69 | 1.05 | ||||
| Pantoea | OTU 314 | 2 | 2 | 0.03 | |||||
| Pseudomonas | OTU 36 | 24 | 20 | 14 | 24 | 5 | 87 | 1.32 | |
| Pseudomonas | OTU 37 | 3 | 3 | 0.05 | |||||
| Pseudomonas | OTU 39 | 2 | 2 | 0.03 | |||||
| Pseudomonas | OTU 46 | 2 | 2 | 0.03 | |||||
| Pseudomonas | OTU 50 | 2 | 2 | 0.03 | |||||
| Pseudomonas | OTU 56 | 1 | 1 | 2 | 0.03 | ||||
| Pseudomonas | OTU 75 | 2 | 2 | 0.03 | |||||
| Pseudomonas | OTU 168 | 1 | 1 | 2 | 0.03 | ||||
| Pseudomonas | OTU 185 | 4 | 4 | 0.06 | |||||
| Pseudomonas | OTU 282 | 2 | 2 | 0.03 | |||||
| Pseudomonas | OTU 383 | 121 | 5 | 9 | 135 | 2.06 | |||
| Pseudomonas | OTU 419 | 1 | 1 | 2 | 0.03 | ||||
| Pseudomonas | OTU 421 | 1 | 1 | 2 | 0.03 | ||||
| Pseudomonas | OTU 600 | 3 | 3 | 0.05 | |||||
| Pseudomonas | OTU 661 | 2 | 2 | 0.03 | |||||
| Stenotrophomonas | OTU 239 | 3 | 21 | 16 | 40 | 0.61 | |||
| Stenotrophomonas | OTU 240 | 1 | 1 | 2 | 0.03 |
In this study, the second most abundant phylum in the maize rhizosphere was Proteobacteria, in which the most predominant genera were Enterobacter and Pseudomonas. In a previous study, E. cloacae was reported to display an endophytic distribution within maize stem and leaf tissues, and exhibited an antagonistic effect against F. verticillioides (Hinton & Bacon, 1995). In this work, this genus was represented by 407 sequences and it was distributed across 3 locations (I, II, and III). Pseudomonas species have been previously described as biocontrol agents (Mendes et al., 2011). Costa et al. (2006) associated the functional and structural diversity of Pseudomonas by matching dominant ribotypes (DGGE) of Pseudomonas spp. in the maize rhizosphere with PCR-DGGE fingerprints of bacterial isolates that display an antagonistic potential against the phytopathogenic bacteria Ralstonia solanacearum. Two abundant OTUs have been previously identified as P. putida (OTU 36; Ps3) and P. fluorescens (OTU 383: Ps42) (Figueroa-López et al., 2016). OTU 36 belongs to the core community of OTUs.
Maize root exudates, such as sugars, organic acids, aromatics, and enzymes interact with soil traits, such as pH, water potential, texture, and nutrient availability, as well as existing microbial populations to promote plant growth and development (Peiffer et al., 2013).
The slight changes on soil physicochemical parameters found in our sampling points may account for differences in microbiota composition between sites (Etesami et al., 2017; Wang et al., 2019).
Although our study provides some insight into the culturable bacterial communities from the maize rhizosphere, a more thorough investigation including culture-independent methods is necessary to resolve which bacterial communities are associated with the maize rhizosphere in agroecosystems.
Since 2009, the viability (95%) of the bacterial isolates from the collection keeps being monitored periodically and the last time the collection viability has been measured in 2019, the viability remains at 89.6%. To the best of our knowledge, this constitutes the largest culturable maize rhizospheric bacterial collection in Mexico. This study also emphasizes the importance of microbial biodiversity conservation through the creation of bacterial collections designed to explore future industrial needs.
Acknowledgements
JDCR and AMFL received Ph.D. fellowships from the Consejo Nacional de Ciencia y Tecnología (Conacyt) (Mexico) and SIP-IPN. To Dr. Brandon Loveall for English proofreading of the manuscript. Financial support for this project was provided by the Fundación Produce Sinaloa (2009-2015) and the Secretaría de Investigación y Posgrado del Instituto Politécnico Nacional (SIP-IPN) (2009-2019).
References
Bressan, W., & Fontes-Figueiredo, J. E. (2007). Efficacy and dose–response relationship in biocontrol of Fusarium disease in maize by Streptomyces spp. European Journal of Plant Pathology, 120, 311–316. https://doi.org/10.1007/s10658-007-9220-y
Bulgarelli, D., Schlaeppi, K., Spaepen, S., van Themaat, E. V. L., & Schulze-Lefert, P. (2013). Structure and functions of the bacterial microbiota of plants. Annual Review of Plant Biology, 64, 807–838. https://doi.org/10.1146/annurev-arplant-050312-120106
Cavaglieri, L. R., Andrés, L., Ibáñez, M., & Etcheverry, M. G. (2005). Rhizobacteria and their potential to control Fusarium verticillioides: effect of maize bacterisation and inoculum density. Antonie van Leeuwenhoek, 87, 179–187. https://doi.org/10.1007/s10482-004-3193-z
Cavaglieri, L., Passone, A., & Etcheverry, M. G. (2004). Screening procedures for selecting rhizobacteria with biocontrol effects upon Fusarium verticillioides growth and fumonisin B1 production. Research in Microbiology, 155, 747–754. https://doi.org/10.1016/j.resmic.2004.06.001
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., McGarrell, D. M., Sun, Y. et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Research, 42, D633–D642. https://doi.org/10.1093/nar/gkt1244
Costa, R., Gomez, N., Peixoto, R. S., Rumjanek, N., Berg, G., Mendonca-Hagler, L. C. et al. (2006). Diversity and antagonistic potential of Pseudomonas spp. associated to the rhizosphere of maize grown in a subtropical organic farm. Soil Biology & Biochemistry, 38, 2434–2447. https//doi.org/10.1016/j.soilbio.2006.03.003
De Man, J. C., Rogosa, M., & Sharpe, M. E. (1960). A medium for cultivation of lactobacilli. Journal of Applied Bacteriology, 23, 130–135. https://doi.org/10.1111/j.1365-2672.1960.tb00
188.x
Douriet-Gámez, N. R., Maldonado-Mendoza, I. E., Ibarra-Laclette, E., Blom, J., & Calderón-Vazquez, C. L. (2018). Genomic analysis of Bacillus sp. strain B25, a biocontrol agent of maize pathogen Fusarium verticillioides. Current Microbiology, 75, 247–255. https://doi.org/10.1007/s00284-
017-1372-1
Etesami, H., Emami, S., & Alikhani, H. A. (2017). Potassium solubilizing bacteria (KSB): Mechanisms, promotion of plant growth, and future prospects A review. Journal of Soil Science and Plant Nutrition, 17, 897–911. https://dx.doi.org/10.4067/S0718-95162017000400005
Figueroa-López, A. M., Cordero-Ramírez, J. D., Martínez-Álvarez, J. C., López-Meyer, M., Lizárraga-Sánchez, G. J., Félix-Gastélum, R. et al. (2016). Rhizospheric bacteria of maize with potential for biocontrol of Fusarium verticillioides. Springer Plus, 5, 330–342. https://doi.org/
10.1186/s40064-016-1780-x
Figueroa-López, A. M., Cordero-Ramírez, J. D., Quiroz-Figueroa, F. R., & Maldonado-Mendoza, I. E. (2014). A high-throughput screening assay to identify bacterial antagonists against Fusarium verticillioides. Journal of Basic Microbiology, 54, S125–S133. https://doi.org/10.1002/jobm.201200594
Figueroa-López, A. M., Leyva-Madrigal, K. Y., Cervantes-Gámez, R. G., Beltrán-Arredondo, L. I., Douriet-Gámez, N. R., Castro-Martínez, C. et al. (2017). Induction of Bacillus cereus chitinases as a response to lysates of Fusarium verticillioides. Romanian Biotechnological Letters ARS Docendi, 22, 12722–12731. https://e-repository.org/rbl/vol.22/iss.4/6.pdf
H. Ayuntamiento de Guasave. Gobierno del Estado de Sinaloa; Secretaría de Desarrollo Social, Medio Ambiente y Pesca; Subsecretaría de Desarrollo Urbano y Ecológico; Dirección de Recursos Naturales y Medio Ambiente. (1998). “La Uba” Cofradía de Tamazula, Guasave. Zona de Preservación Ecológica de Centro de Población. Guasave, Sinaloa.
Haas, D., Keel, C., & Reimmann, C. (2002). Signal transduction in plant-beneficial rhizobacteria with biocontrol properties. Antonie van Leeuwenhoek, 81, 385–395. https://doi.org/10.
1023/A:1020549019981
Hinton, D. M., & Bacon, C. W. (1995). Enterobacter cloacae is an endophytic symbiont of corn Mycophathologia, 129, 117–125. https://doi.org/10.1007/bf01103471
Ibarra-Galeana, J. A., Castro-Martínez, C., Fierro-Coronado, R. A., Armenta-Bojórquez, A. D., & Maldonado-Mendoza, I. E. (2017). Characterization of phosphate-solubilizing bacteria exhibiting the potential for growth promotion and phosphorus nutrition improvement in maize (Zea mays L.) in calcareous soils of Sinaloa, Mexico. Annals of Microbiology, 67, 801–811. https://doi.org/10.1007/s13213-017-1308-9
Kumar, S., Stecher, G., Li, M., Knyaz, C., & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. https://doi.org/10.1093/molbev/msy096
Lennon, J. T., & Jones, S. E. (2011). Microbial seed banks: the ecological and evolutionary implications of dormancy. Nature Reviews Microbiology, 9, 119–130. https://doi.org/
10.1038/nrmicro2504
Li, X., Rui, J., Xiong, J., Li, J., He, Z., Zhou, J. et al. (2014). Functional potential of soil microbial communities in the maize rhizosphere. Plos One, 9, e112609. https://doi.org/10.1371/journal.pone.0112609
Liu, X., Wang, Y., Liu, Y., Chen, H., & Hu, Y. (2020). Response of bacterial and fungal soil communities to chinese fir (Cunninghamia lanceolate) long-term monoculture plantations. Frontiers in Microbiology, 11, 181. https://doi.org/10.3389/fmicb.2020.00181
Lizárraga-Sánchez, G. J., Leyva-Madrigal, K. Y., Sánchez-Peña, P., Quiroz-Figueroa, F. R., & Maldonado-Mendoza, I. E. (2015). Bacillus cereus sensu lato strain B25 controls maize stalk and ear rot in Sinaloa, Mexico. Field Crops Research, 176, 11–21. https://doi.org/10.1016/j.fcr.2015.02.015
López-Bucio, J., Campos-Cuevas, J. C., Hernández-Calderón, E., Velásquez-Becerra, C., Farías-Rodríguez, R., Macías-Rodríguez, L. I. et al. (2007). Bacillus megaterium rhizobac-
teria promote growth and alter root-system architecture through an auxin- and ethylene-independent signaling mechanism in Arabidopsis thaliana. Molecular Plant Microbe Interactions, 20, 207–217. https://doi.org/10.1094/MPMI-20-2-0207
Lu, J. J., Perng, C. L., Lee, S. Y., & Wan, C. C. (2000). Use of PCR with universal primers and restriction endonuclease digestions for detection and identification of common bacterial pathogens in cerebrospinal fluid. Journal of Clinical Microbiolology, 38, 2076–2080. https://doi.org/10.1128/JCM.38.6.2076-2080.2000
Lucon, C. M. M., Guzzo, S. D., De Jesus, C. O., Pascholati, S. F., & De Goes, A. (2010). Postharvest harpin or Bacillus thuringiensis treatments suppress citrus black spot in ‘Valencia’ oranges. Crop Protection, 29, 766–772. https://doi.org/10.1016/j.cropro.2010.02.018
Mendes, R., Kruijt, M., de Bruijn, I., Dekkers, E., van der Voort, M., Schneider, J. H. et al. (2011). Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science, 332, 1097–1100. https://doi.org/10.1126/science.1203980
Morales-Ruiz, E., Priego-Rivera, R., Figueroa-López, A. M., Cazares-Álvarez, J. E., & Maldonado-Mendoza, I. E. (2021) Biochemical characterization of two chitinases from Bacillus cereus sensu lato B25 with antifungal activity against Fusarium verticillioides P03. FEMS Microbiology Letters, 368, fnaa218. https://doi.org/10.1093/femsle/fnaa218
Nagórska, K., Bikowski, M., & Obuchowski, M. (2007). Multicellular behaviour and production of a wide variety of toxic substances support usage of Bacillus subtilis as a powerful biocontrol agent. Acta Biochimica Polonica, 54, 495–508. https://doi.org/10.18388/ABP.2007_3224
Olsen, S. R., Cole, C. V., Watanabe, F. S., & Dean, L. A. (1954). Estimation of available phosphorus in soils by extraction with sodium bicarbonate. USDA Circular, 939, 1–19. Gov. Printing Office, Washington D.C., USA.
Omotayo, O. P., Igiehon, O. N., & Babalola, O. O. (2021). Metagenomic study of the community structure and functional potentials in maize rhizosphere microbiome: Elucidation of mechanisms behind the improvement in plants under normal and stress conditions. Sustainability, 13, 8079. https://doi.org/10.3390/su13148079
Pasarell, L., & McGinnis, M. R. (1992). Viability of fungal cultures maintained at -70 oC. Journal of Clinical Microbiology, 30, 1000-1004. https://doi.org/10.1128/jcm.30.4.1000-1004.1992
Peiffer, J. A., Spor, A., Koren, O., Jin, Z., Tringer, S. G., Dangl, J. L. et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proceedings of the National Academy of Sciences, 110, 6548–6553. https://doi.org/10.1073/pnas.1302837110
Pereira, P., Ibáñez, F., Rosenblueth, M., Etcheverry, M., & Martínez-Romero, E. (2011). Analysis of the bacterial diversity associated with the roots of maize (Zea mays L.) through culture-dependent and culture-independent methods. International Scholarly Research Notices Ecology, 2011, 938546. https://doi.org/10.5402/2011/938546
Saleem, M., Hu, J., & Jousset, A. (2019). More than the sum of its parts: Microbiome biodiversity as a driver of plant growth and soil health. Annual Review of Ecology, Evolution, and Systematics, 50, 145–168. https://doi.org/10.1146/annurev-ecolsys-110617-062605
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B. et al. (2009). Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75, 7537–7541. https://doi.org/10.1128/AEM.01541-09
Shi, T., Reeves, R. H., Gilichinsky, D. A., & Friedmann, E. I. (1997). Characterization of viable bacteria from Siberian permafrost by 16S rDNA sequencing. Microbial Ecology, 33, 169–179. https://doi.org/10.1007/s002489900019
SIAP (Servicio de Información Agroalimentaria y Pesquera). (2020). Disponible en: https://nube.siap.gob.mx/cierreagricola/
Sun, D. L., Jiang, X., Wu, Q. L., & Zhou, N. Y. (2013). Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Applied and Environmental Microbiology, 79, 5962–5969. https://doi.org/10.1128/AEM.01282-13
Tamura, K., & Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution, 10, 512–526. https://doi.org/10.1093/oxfordjournals.molbev.a040023
Thompson, J. D., Higgins, D. G., & Gibson, T. J. (1994). CLUSTAL W improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research, 22, 4673–4680. https://doi.org/10.1093/nar/22.22.4673
Tilak, K., & Reddy, B. (2006). Bacillus cereus and B. circulans – novel inoculants for crops. Current Science, 90, 642–644. www.jstor.org/stable/24089111
Trejo-Aguilar, D., Lara-Capistrán, L., Maldonado-Mendoza, I. E., Zulueta-Rodríguez, R., Sangabriel-Conde, W., et al. (2013) Loss of arbuscular mycorrhizal fungal diversity in trap cultures during long-term subculturing. IMA Fungus, 4, 161–167. https://doi.org/10.5598/imafungus.2013.04.
02.01
Walkley, A., & Black, I. A. (1934). An examination of Degtjareff method for determining soil organic matter and a proposed modification of the chromic acid titration method. Soil Science, 37, 29–38. http://dx.doi.org/10.1097/00010694-193401000-00003
Wang, C., Zhou, X., Guo, D., Zhao, J., Yan, L., Feng, G. et al. (2019). Soil pH is the primary factor driving the distribution and function of microorganisms in farmland soils in northeastern China. Annals of Microbiology, 69, 1461–1473. https://doi.org/10.1007/s13213-019-01529-9
Yang, Y., Wang, N., Guo, X., Zhang, Y., & Ye, B. (2017). Comparative analysis of bacterial community structure in the rhizosphere of maize by high-throughput pyrosequencing. Plos One, 12, e0178425. https://doi.org/10.1371/journal.pone.0178425
Zhang, X., Zhang, R., Gao, J., Wang, X., Fan, F., Ma, X. et al. (2017). Thirty-one years of rice-rice-green manure rotations shape the rhizosphere microbial community and enrich beneficial bacteria. Soil Biology and Biochemistry, 104, 208–217. https://doi.org/10.1016/j.soilbio.2016.10.023


