
Francisco J. Vázquez-Ponce a, b, Giovani Hernández-Canchola c, Andrea R. Jiménez-Marín d y Lázaro Guevara a, *
a Universidad Nacional Autónoma de México, Instituto de Biología, Departamento de Zoología, Colección Nacional de Mamíferos, Apartado postal 70-153, 04510 Ciudad de México, México
b Universidad Veracruzana, Facultad de Ciencias Biológicas y Agropecuarias, Camino Peñuela-Amatlán s/n Peñuela, 94500 Amatlán de los Reyes, Veracruz, México
c Lousiana State University, Museum of Natural Science, 119 Foster Hall, 70803, Baton Rouge, LA, EUA
d Universidad Nacional Autónoma de México, Instituto de Biología, Departamento de Zoología, Laboratorio de Biología Molecular, Apartado postal 70-153, 04510 Ciudad de México, México
*Autor para correspondencia: llg@ib.unam.mx (L. Guevara)
Recibido: 8 octubre 2020; aceptado: 22 febrero 2021
Resumen
Las musarañas del grupo Cryptotis mexicanus (Mammalia, Soricidae) están estrechamente asociadas con el bosque húmedo de montaña al norte del Neotrópico. Dentro de este grupo, los estudios filogenéticos han definido un grupo monofilético compuesto por 3 especies: C. nelsoni como hermana del clado C. obscurus – C. mexicanus. El patrón conocido de variación morfológica y las estimaciones de distribución potencial sugieren que su evolución está asociada con los cambios altitudinales inducidos por los últimos ciclos glaciales-interglaciales. En este estudio analizamos 70 secuencias mitocondriales del gen citocromo b para este clado mediante análisis filogeográficos y filogenéticos, esto para evaluar si los valles, cañones y planicies, a lo largo de la distribución de este grupo podrían estar promoviendo su estructura y diferenciación genética. Los resultados indican 4 grupos genéticos, parcialmente congruentes con estudios previos; uno de ellos corresponde a C. nelsoni, mientras que los otros 3 aparecen dentro del clado C. obscurus – C. mexicanus. La divergencia genética entre los 4 grupos sugiere la influencia de algunas barreras geográficas, desde valles y cañones hasta volcanes, que a través de los últimos ciclos glaciales-interglaciales podrían haber conformado el patrón actual de variación genética. Nuestros hallazgos sugieren que la taxonomía entre C. obscurus y C. mexicanus aún no está resuelta.
Palabras clave: Bosque de niebla; ADN mitocondrial; Eulipotyphla; Filogeografía; México
© 2021 Universidad Nacional Autónoma de México, Instituto de Biología. Este es un artículo Open Access bajo la licencia CC BY-NC-ND
Genetic divergence in shrews (Mammalia: Soricidae) of humid montane forests north of the Neotropics
Abstract
Shrews of the Cryptotis mexicanus group (Mammalia, Soricidae) are closely associated with the humid mountain forest north of the Neotropics. Within this group, phylogenetic studies have defined a monophyletic group composed of 3 species: C. nelsoni as sister of the clade C. obscurus – C. mexicanus. The pattern of morphological variation and estimates of potential distribution suggests that its evolution is tightly associated with the altitudinal changes induced by the last glacial-interglacial cycles. In this study, we analyzed 70 mitochondrial sequences of the Cytochrome b for this clade using phylogeographic and phylogenetic analyses to test whether the valleys, canyons, and plains along the distribution of this group could be promoting its genetic structure and differentiation. The results indicate 4 genetic groups, partially congruent with previous studies; one corresponds to C. nelsoni, while the other 3 appear within the clade C. obscurus – C. mexicanus. The genetic divergence observed among the 4 groups suggests the influence of some geographical barriers, from valleys and canyons to volcanoes, which through the last glacial-interglacial cycles could have shaped the current genetic pattern variation. Our findings also suggest that the taxonomy between C. obscurus and C. mexicanus is not yet resolved.
Keywords: Cloud forest; Mitochondrial DNA; Eulipotyphla; Phylogeography; Mexico
© 2021 Universidad Nacional Autónoma de México, Instituto de Biología. This is an open access article under the CC BY-NC-ND license
Introducción
Las musarañas (Mammalia, Soricidae) son los mamíferos de menor tamaño en el mundo. Debido a sus demandas energéticas, son depredadores voraces de artrópodos y anélidos, pero también son presas comunes de aves rapaces, serpientes y mamíferos carnívoros (Churchfield, 1990). Con cerca de 440 especies agrupadas en 26 géneros, representan a una de las familias de mamíferos con mayor distribución geográfica en el planeta, habitando todos los ecosistemas terrestres (Burgin et al., 2018). Su capacidad de dispersión relativamente limitada, tiempo de generación corto y asociaciones estrechas con hábitats particulares, han contribuido a que las musarañas reciban una atención especial para evaluar el papel que han jugado el cambio climático del pasado y la orografía en la conformación de los patrones de variabilidad genética actual y los procesos de divergencia de linajes. Lo anterior ha sido particularmente importante en regiones templadas, boreales y paleotropicales (Giarla y Esselstyn, 2015; Hope et al., 2020; Jacquet et al., 2013; Vega et al., 2020; Vuilleumier y Fontanillas, 2007). En el Neotrópico, la situación es diferente. El acervo relativamente limitado tanto de ejemplares en colecciones biológicas como de muestras para análisis genéticos, así como el número reducido de taxónomos, han impedido un avance sustancial en el conocimiento sobre la evolución y biogeografía del grupo (Lorenzo et al., 2019; Noguera-Urbano et al., 2019).
Por su diversidad y distribución, las musarañas de orejas cortas (género Cryptotis) son el representante principal de musarañas neotropicales y el único género que habita en Norte y Sudamérica. Con pocas excepciones, las especies están restringidas a ecosistemas de media y alta montaña, en donde se combinan condiciones de humedad y precipitación alta con temperatura baja (Guevara et al., 2015; Noguera-Urbano et al., 2019; Woodman et al., 2012). Dentro del género, el grupo de especies Cryptotis mexicanus comprende a musarañas exclusivas del norte del Neotrópico y estrechamente asociadas al bosque húmedo de montaña (o simplemente bosque de niebla), entre los 500 y los 3,000 m snm (Guevara et al., 2015). Esta agrupación de especies se caracteriza por poseer extremidades anteriores grandes que han sido interpretadas como adaptaciones para explotar hábitos semifosoriales (Guevara, 2017; Woodman, 2011).
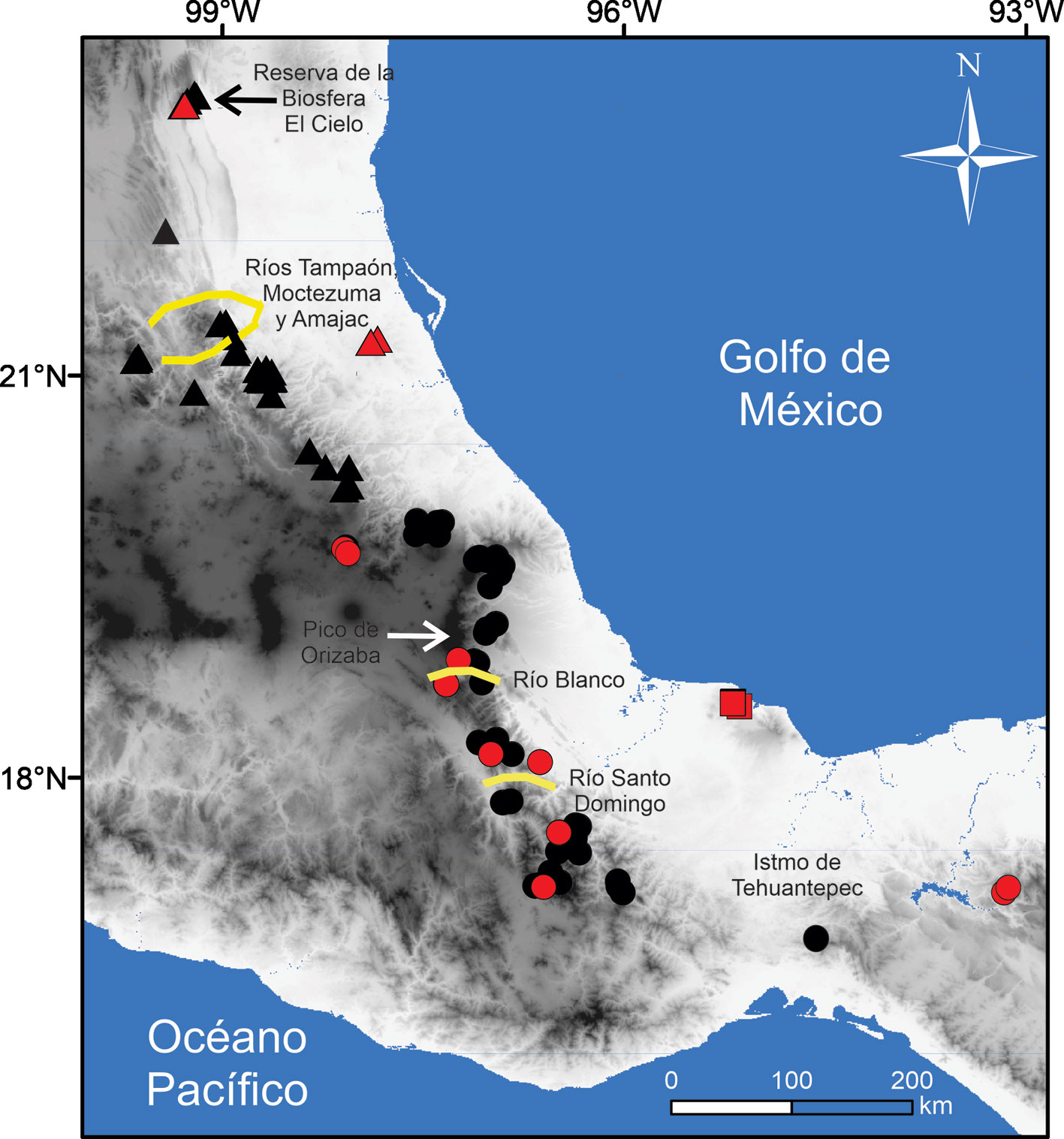
En el grupo C. mexicanus, las relaciones filogenéticas muestran a un clado distribuido en el extremo boreal compuesto por 3 especies: C. mexicanus (Coues, 1877) y C. obscurus (Merriam, 1895) cercanamente relacionadas y hermanas de C. nelsoni (Merriam, 1895) (Guevara y Cervantes, 2014; He et al., 2015). Cryptotis obscurus se distribuye en las provincias biogeográficas de Sierra Madre Oriental y el norte de la Veracruzana; C. mexicanus al este de la Faja Volcánica Transmexicana, noreste de la Sierra Madre del Sur y en las Tierras Altas de Chiapas; mientras que C. nelsoni es endémica de la Sierra de Los Tuxtlas, al este de la provincia Veracruzana (fig. 1). Dada su afinidad con el bosque de niebla de estas provincias, se ha especulado que la evolución de estas musarañas está sincronizada con la intrincada historia de expansión, conectividad y fragmentación de dicho ecosistema a través de los últimos ciclos glaciales-interglaciales del Cuaternario (Choate, 1970; Guevara y Sánchez-Cordero, 2018a).
Las glaciaciones, por un lado, habrían ocasionado el descenso en elevación y la expansión de los bosques de niebla (Caballero et al., 2010), por lo que las tierras bajas servirían como puentes para permitir la conectividad de poblaciones; mientras que durante los interglaciales (como en el presente), estas zonas bajas fungen como barreras que aíslan a las poblaciones al no poseer condiciones idóneas para el desarrollo del bosque de niebla (Ramírez-Barahona y Eguiarte, 2013). En específico, los valles y cañones que conducen a los ríos Tampaón, Moctezuma, Amajac al norte de la Sierra Madre Oriental, a los ríos Blanco y Santo Domingo al norte de la Sierra Madre del Sur, la planicie hacia la costa del Golfo de México y el Istmo de Tehuantepec, podrían actuar como barreras y corredores que aíslan y conectan a las poblaciones de estas especies durante las oscilaciones climáticas del Cuaternario (Guevara y Sánchez-Cordero, 2018a).
Una revisión taxonómica reciente no encontró una diferenciación morfológica ni ecológica significativa entre C. obscurus y C. mexicanus, lo cual podría ser consecuencia de un proceso de divergencia reciente o una conectividad frecuente entre las poblaciones durante los ciclos glaciales-interglaciales. Esto ha dejado dudas sobre el estado taxonómico de ambas especies (Guevara y Sánchez-Cordero, 2018a). Por el otro lado, la diferenciación morfológica notable de C. nelsoni podría deberse a su distribución restringida en el sistema montañoso de la Sierra de Los Tuxtlas, donde la especie se encuentra aislada y probablemente sin flujo con el resto de las especies por un tiempo largo (Guevara y Sánchez-Cordero, 2018b; Woodman y Timm, 1999), incluso durante la expansión del bosque de niebla en los eventos glaciales más recientes (Guevara, 2020).
Para poner a prueba si los valles y cañones mencionados anteriormente podrían estar promoviendo la divergencia dentro del clado C. mexicanus, C. obscurus y C. nelsoni, sería crucial contar con información genética. Afortunadamente, en los últimos años, el trabajo de campo dirigido a la recolecta de musarañas ha permitido incrementar la representación geográfica y el almacenamiento de muestras para estudios genéticos (Guevara y Sánchez-Cordero, 2018a, b; Mayén-Zaragoza et al., 2019). Por lo anterior, en este estudio usamos secuencias del gen mitocondrial Citocromo b para evaluar la estructura genética y la diferenciación dentro del clado conformado por estas 3 especies. Esperamos que este primer estudio filogeográfico de musarañas neotropicales no solo contribuya a comprender la historia biogeográfica de este clado y otras especies del bosque de niebla en la norte del Neotrópico, sino que repercuta en el conocimiento taxonómico del grupo y su conservación.
Materiales y métodos
Los ejemplares de referencia se encuentran depositados en las siguientes colecciones científicas de México (apéndice): Colección Nacional de Mamíferos (CNMA), Ciudad de México; Museo de Zoología ‘Alfonso L. Herrera’ (MZFC), Ciudad de México; y Colección Mastozoológica de El Colegio de la Frontera Sur (ECO-SC-M), San Cristóbal de las Casas. Se extrajo DNA desde tejido con un kit comercial DNeasy® Tissue Qiagen, y la amplificación del gen mitocondrial Citocromo b (Citb) por medio de la Reacción en Cadena de la Polimerasa (PCR, por su siglas en inglés) usando el par de oligonucleótidos MVZ05 (5′-CGAAGCTTGATATGAAAAACCATCGTTG; Smith y Patton, 1999) y H15915 (5′-AACTGCAGTCATCTCCGGTTTACAAGAC; Irwin et al., 1991). El volumen final de reacción fue de 15 µl utilizando el buffer de la enzima en concentración 1X, 0.16 µM del cebador MVZ05, 0.16 µM del cebador H15915, 0.4 mM de cada dNTP, 0.6 U de Taq Polimerasa MyTaqTM Bioline y una concentración entre 50-265 ng/µl de DNA.
El programa de la PCR incluyó 3 min de desnaturalización inicial a 95 °C; seguidos de 35 ciclos que incluyeron 30 segundos de desnaturalización a 95 °C, un min de alineamiento a 49 °C, y 2 min de extensión a 72 °C. Se visualizó el producto de PCR utilizando 3 µl mediante electroforesis en geles de agarosa al 1.5%. Se utilizó el kit de purificación Montage PCR de Millipore y el kit de secuenciación ABI PRISM Big Dye Terminator en un secuenciador ABI 3730XL (Applied Biosystems, Foster City, CA, USA). Se obtuvieron 46 secuencias genéticas del clado conformado por C. mexicanus (29), C. obscurus (7) y C. nelsoni (10). Se realizó la edición y limpieza de las secuencias por medio de una inspección visual con el programa MEGA X (Kumar et al., 2018). Además, se obtuvieron 24 secuencias de la base de datos GenBank (C. mexicanus [9], C. obscurus [2] y C. nelsoni [5] y 8 utilizadas como grupos externos (apéndice).
Se identificó la distribución espacial de la variabilidad genética mediante el paquete Geneland ® v. 4.0.8 en R® v3.4.4, el cual localiza discontinuidades genéticas entre las muestras (Guillot et al., 2005). Para ello, se utilizó el modelo haploide, un modelo de frecuencias alélicas no correlacionadas y un modelo espacial verdadero. También se usaron las cadenas de Markov Monte-Carlo con 6 corridas independientes de 5,000,000 iteraciones cada una, muestreando datos cada 1,000. Aplicamos un descarte (“burn-in”) del 25%, comparamos las corridas independientes y seleccionamos el análisis con mayor probabilidad posterior.
Se construyó una red de haplotipos para establecer la relación entre todos los haplotipos mediante una red de unión de medianas (“median-joining”) usando el programa Network v10.0 (Fluxus Technology Ltd., 2015). Calculamos la diversidad haplotídica, nucleotídica, así como el número de sitios polimórficos en el programa DNAsp v6.12.02 (Librado y Rozas, 2009).
Para medir la diferencia genética en el Citb entre los grupos identificados en el análisis de agrupación espacial bayesiana, se calcularon las distancias genéticas utilizando el programa MEGA X (Kumar et al., 2018). Para ello, se agruparon las secuencias en los grupos identificados y se ejecutó un análisis de distancia neta entre el promedio de los grupos. Los parámetros definidos fueron 10,000 repeticiones de bootstrap mediante el modelo p-distance (Starcová et al., 2016). Asimismo, se analizó la distribución de la variabilidad genética entre los grupos definidos previamente. Para ello, se realizó un análisis de varianza molecular (Amova) implementando 16,000 permutaciones para el cómputo de una matriz de distancias, bajo el modelo de proporción de diferencias, usando el programa Arlequin (Excoffier y Lischer, 2010).
Se llevó a cabo un análisis de inferencia bayesiana para determinar las relaciones filogenéticas entre los grupos identificados en los análisis previos. Para estimar el mejor modelo evolutivo de sustitución nucleotídica utilizado para la inferencia bayesiana, utilizamos el software Smart model selection (SMS) en el entorno de PhyML (Anisimova y Gascuel, 2006; Lefort et al., 2017)AIC. Según el criterio de información bayesiano (BIC), el mejor modelo de sustitución nucleotídica para los datos fue el modelo HKY85+G. La inferencia bayesiana se realizó en el programa Mr.Bayes v3.2.7 (Ronquist et al., 2012). Se realizaron 2 corridas con 15 millones de generaciones e implementando 8 Cadenas de Markov Monte Carlo (MCMC; 7 cadenas calientes y una fría). Se muestrearon cada 500 generaciones, descartando el primer 25% de la muestra (burn-in). Finalmente, se generó el árbol de mayoría con los valores de probabilidad posterior para cada clado. Para analizar y visualizar los resultados, se usaron los programas Tracer v1.7.1 y FigTree v1.4.4 (Rambaut et al., 2018).
Resultados
El análisis de agrupación espacial reveló 4 grupos genéticos dentro del clado conformado por C. mexicanus, C. obscurus y C. nelsoni (fig. 2). La probabilidad posterior de que existieran más de 4 grupos según el Citb fue extremadamente baja. El primer grupo (en adelante denominado NORTE) incluye a todas las muestras pertenecientes a C. obscurus y a los haplotipos previamente identificados como C. mexicanus del centro del país y de la Sierra de Otontepec, Veracruz, al este de la Sierra Madre Oriental (fig. 2A). El segundo grupo (CENTRO; fig. 2B) está formado por haplotipos de C. mexicanus desde los alrededores del volcán Pico de Orizaba, Veracruz hasta el norte del río Santo Domingo, Oaxaca. El tercer grupo (SUR) está compuesto por todas las muestras de C. mexicanus al sur del río Santo Domingo, incluyendo a las 2 secuencias al este del istmo de Tehuantepec, Oaxaca (fig. 2C). Finalmente, el cuarto grupo (LOS TUXTLAS) está formado exclusivamente por los haplotipos correspondientes a C. nelsoni, en el sistema montañoso aislado de Los Tuxtlas (fig. 2D). Los valores máximos de probabilidad posterior para la pertenencia poblacional de los haplotipos fueron cercanos a 50%, mientras que la probabilidad posterior de que los haplotipos de un grupo pertenecieran a cualquiera de las otras 3 agrupaciones fue considerablemente menor.
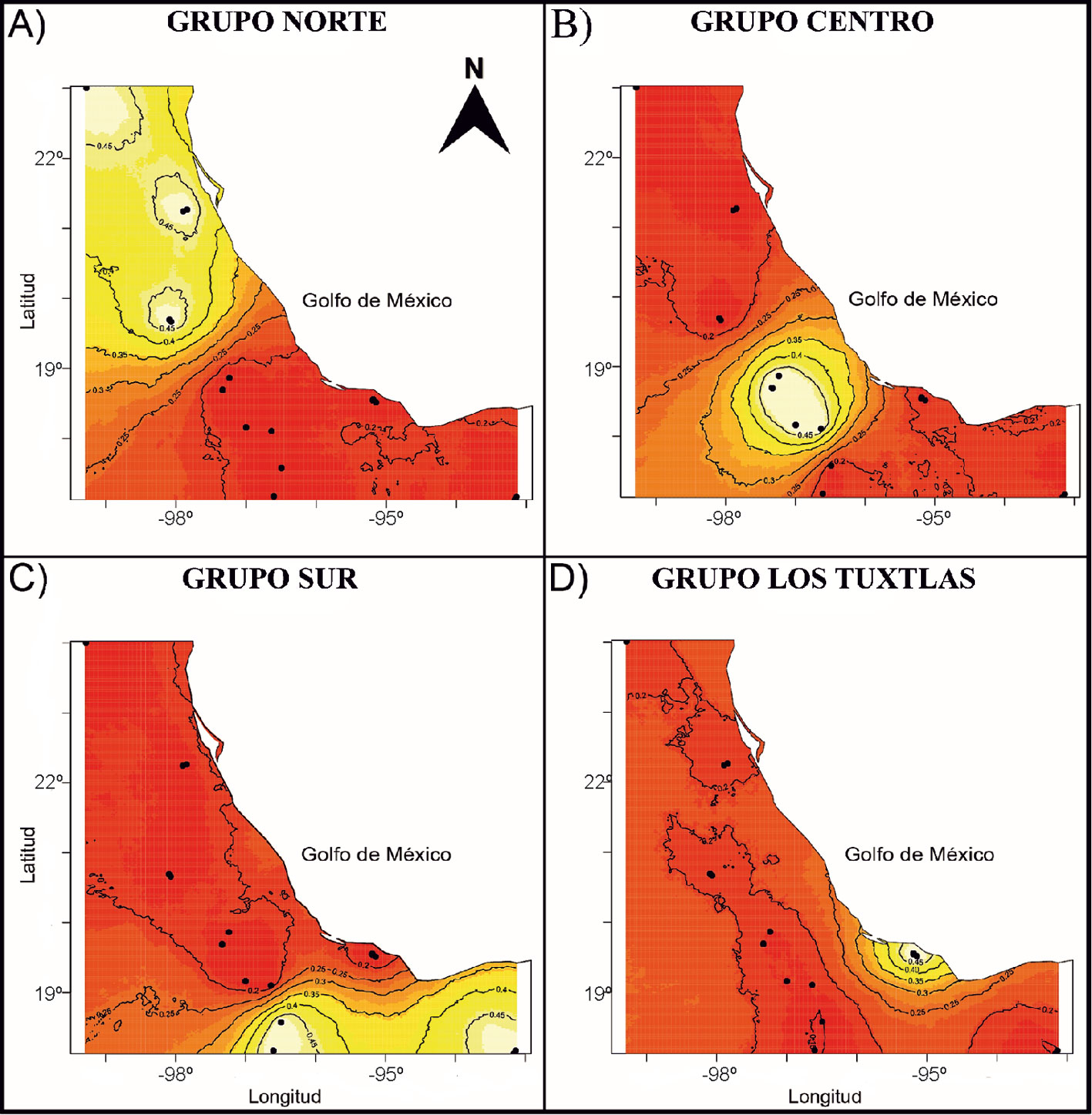
Se obtuvieron 42 haplotipos en la muestra total (Hd: 0.9672; Pi: 0.0469). El número de sitios polimórficos fue de 211 en las 1,050 pb (20.11%). La red mostró haplogrupos similares a los resultados obtenidos por el análisis de agrupación espacial bayesiana, donde el grupo NORTE se formó con 19 secuencias distribuidas en 15 haplotipos. A su vez, el grupo CENTRO estuvo compuesto por 19 secuencias con 12 haplotipos y el SUR por igual número de secuencias y 8 haplotipos. Ambos están formados únicamente por haplotipos de C. mexicanus. En el caso de LOS TUXTLAS, se obtuvieron 7 haplotipos de C. nelsoni en las 16 secuencias genéticas examinadas.
En el grupo NORTE, se observó la fragmentación entre diferentes regiones geográficas incluidas en el estudio (fig. 3). Este grupo requirió al menos 48 pasos mutacionales respecto al grupo SUR y 62 pasos mutacionales respecto al grupo de LOS TUXTLAS. Para el caso del grupo SUR, los 2 haplotipos más distantes al resto del mismo grupo corresponden a aquellos localizados al este del istmo de Tehuantepec, requiriendo al menos 33 pasos mutacionales al nodo más próximo. Por otro lado, al menos 42 pasos mutacionales separan al grupo CENTRO del grupo SUR. En el caso del grupo de LOS TUXTLAS (C. nelsoni), los pocos haplotipos se mostraron cercanos entre sí y más relacionados con el grupo NORTE.
El grupo genéticamente más distante del resto fue el de LOS TUXTLAS (> 5.1%). Dentro del par de especies C. mexicanus y C. obscurus, la mayor distancia resultó entre la población NORTE y CENTRO (> 4.4%), mientras que los grupos más cercanos genéticamente son el grupo CENTRO y el grupo SUR (2.4 %). El análisis de varianza molecular mostró que más de 70% de la variación genética está entre las poblaciones y no dentro de ellas. Asimismo, el análisis reveló que los valores de diferenciación entre los 4 grupos resultaron significativos (tabla 1).
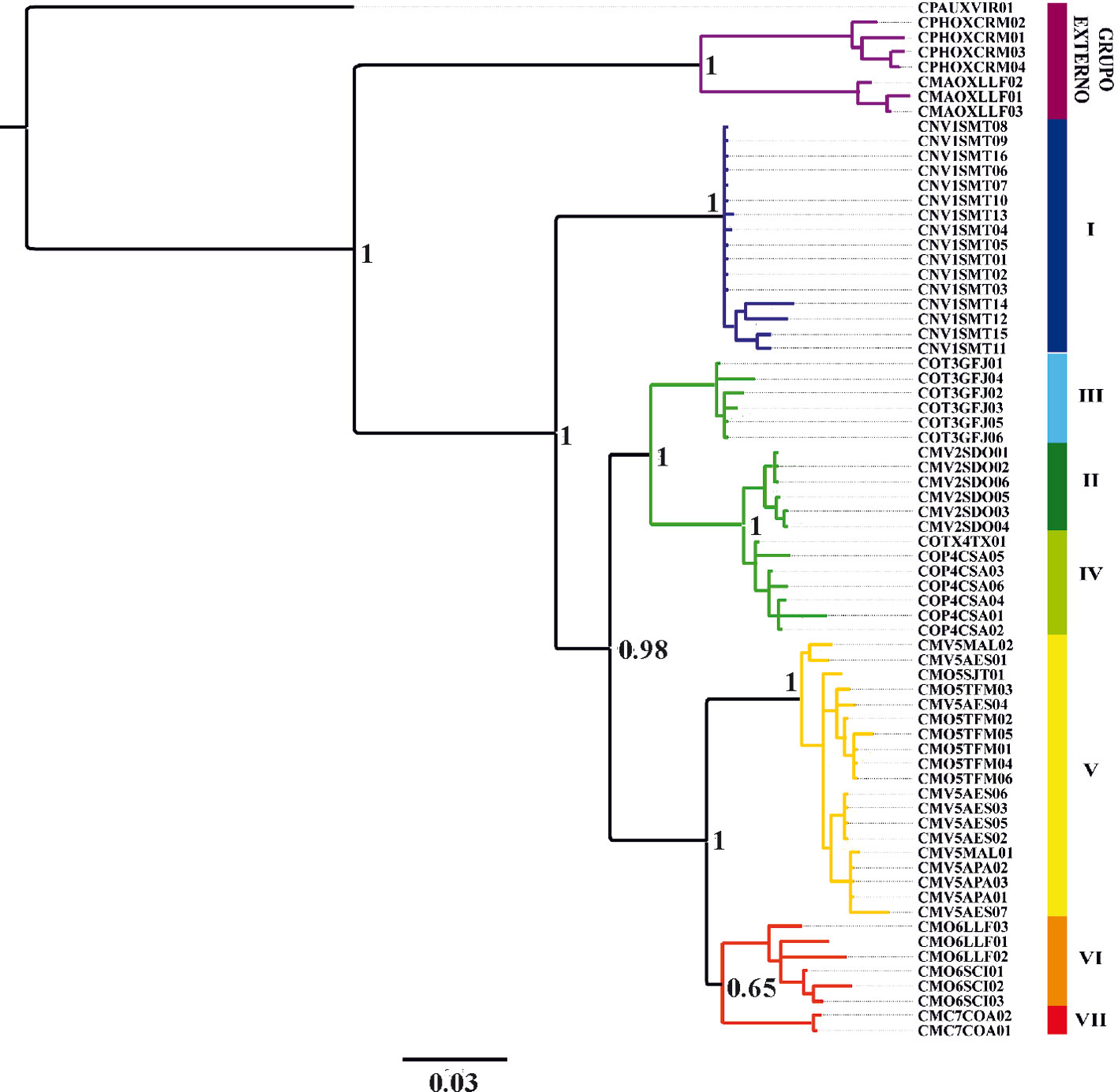
Las frecuencias de cada base nitrogenada en los 1,050 pb examinados se distribuyeron bajo los siguientes porcentajes: 29.3% (T), 29.3% (C), 27.6% (A) y 13.8% (G). El árbol consenso de la inferencia bayesiana mostró resultados altamente congruentes con los resultados de agrupación espacial bayesiana y la red de haplotipos (fig. 4). El grupo de LOS TUXTLAS es un clado bien diferenciado y apoyado (1.0), formado exclusivamente por los haplotipos de C. nelsoni. A su vez, este es grupo hermano del clado que incluye a todas las muestras previamente identificadas como C. mexicanus y C. obscurus. El grupo NORTE se conformó con todos los haplotipos de C. obscurus, además de los de C. mexicanus del centro de México y de la sierra de Otontepec, con alto valor de apoyo (1.0). Este grupo es hermano del clado que contiene a los grupos CENTRO y SUR con valor de apoyo alto (1.0); sin embargo, la monofilia del grupo SUR está débilmente apoyada (0.65).
Discusión
La distribución actual en el espacio geográfico y la divergencia genética entre los 4 grupos dentro del clado integrado por Cryptotis mexicanus, C. obscurus y C. nelsoni sugieren una serie de eventos de expansión, contracción y fragmentación geográfica como consecuencia, probablemente, de las fluctuaciones climáticas a lo largo del Pleistoceno tardío (Ramírez-Barahona y Eguiarte, 2013). Las oscilaciones climáticas y la orografía intrincada del norte del Neotrópico habrían favorecido periodos alternados de conectividad y aislamiento poblacional, dando como resultado el patrón de variabilidad genética observado en estas musarañas (Barber y Klicka, 2010). Si bien ésto no es nuevo y está de acuerdo con lo encontrado en diversos estudios previos (e.g., Guevara, 2020; Ornelas et al., 2010, 2013), el presente estudio destaca el impacto diferencial de las barreras geográficas a lo largo del área de estudio.
Tabla 1
Análisis de varianza molecular (Amova) de la estructura poblacional del complejo C. mexicanus, C. obscurus y C. nelsoni. El porcentaje de variación indica en qué nivel de la estructura poblacional del complejo se encuentra el mayor número de diferencia en la variación molecular.
|
Fuente de la variación |
d.f |
Suma de |
Componentes de varianza |
Porcentaje de variación |
Índices Φ |
* (p < 0.05) |
|
Entre grupos |
3 |
1,136.580 |
21.83162 Va |
70.54 |
0.70537 |
* |
|
Entre las poblaciones dentro de los grupos |
9 |
233.529 |
6.49965 Vb |
21.00 |
0.71277 |
* |
|
Dentro de las poblaciones |
49 |
128.343 |
2.61925 Vc |
8.46 |
0.91537 |
* |

El grupo NORTE se observa como una agrupación cohesiva, pero fragmentada geográficamente y con estructura genética interna. La población en el extremo boreal, aislada en la Reserva de la Biosfera El Cielo, Tamaulipas, muestra una estrecha relación con las muestras al sur. Algunas zonas bajas como las subcuencas de los ríos Tampaón, Moctezuma y Amajac en los estados de Hidalgo y Querétaro, afluentes del Pánuco, parecen restringir de manera intermitente el flujo genético entre ambas regiones, evidenciando el papel de barreras ribereñas en musarañas (Jacquet et al., 2013; tabla 1, figs. 2, 3). Al noreste, las tierras bajas húmedas de la provincia Veracruzana, a lo largo de la costa del golfo de México, han mantenido en aislamiento a la Sierra de Otontepec, un grupo de montañas aisladas de hasta 1,300 m de altitud (Mayen-Zaragoza et al., 2019). A pesar de este notable aislamiento actual, las muestras de Otontepec también están estrechamente relacionadas con el centro y norte de la Sierra Madre Oriental.
En general, la agrupación NORTE es biogeográficamente interesante, ya que apoya la predicción del desplazamiento del bosque de niebla hacia la vertiente oriental de la Sierra Madre Oriental durante el Último Máximo Glacial y se acentúa con la deglaciación en el evento 1 de Heinrich (hace entre 17,000 y 15,000 años; Guevara, 2019). Parece prudente sugerir que las poblaciones dentro del grupo NORTE estuvieron en contacto durante las fases glaciales, siguiendo la expansión del bosque de niebla, pero fragmentándose durante las fases interglaciales.
Con respecto al límite meridional del grupo NORTE, la evidencia genética indica que la diferenciación entre los grupos NORTE y CENTRO es alta y significativa (tabla 1, fig. 4). Con base en el análisis de discontinuidades genéticas, es posible inferir que las zonas más elevadas y frías de la Faja Volcánica Transmexicana estén actuando como una barrera para estas musarañas (e.g., la cara norte del volcán Pico de Orizaba), y no así el cañón del río Blanco como había sido sugerido por Guevara y Sánchez-Cordero (2018a). De ser así, el límite entre ambos grupos apoya lo sugerido originalmente por Choate (1970), quien mencionó que alrededor de los estados de Hidalgo, Puebla y Veracruz podría estar una zona de contacto entre C. obscurus y C. mexicanus.
El cañón del río Santo Domingo (ca. 450 m snm), por su parte, estaría actuando como una barrera intermitente o reciente entre los grupos CENTRO y SUR. La divergencia genética relativamente baja entre ambos grupos, la monofilia débilmente apoyada del grupo SUR y la alta similitud morfológica (Guevara y Sánchez-Cordero, 2018a), sugieren un efecto tenue del cañón del río Santo Domingo en la divergencia de ambos grupos. El río Santo Domingo parece haber tenido un impacto en la diversificación de algunos pequeños mamíferos, tal como roedores de los géneros Habromys (Carleton et al., 2002) y Megadontomys (Vallejo y González-Cózatl, 2012), pero no tan fuerte para el género Handleyomys (Almendra et al., 2014). Los efectos diferenciales entre especies podrían deberse a los atributos de cada taxón, tal como el tamaño corporal, hábitos y vagilidad.
Al sureste, el istmo de Tehuantepec es una franja de tierras bajas de 250 km de ancho en su punto más angosto, con algunos lomeríos de baja altitud (ca. 250 m snm) (Barrier et al., 1998; Ornelas et al., 2013). Históricamente, se ha sugerido que el istmo es una barrera geográfica importante para la divergencia de diversos grupos taxonómicos, incluyendo las musarañas (Choate, 1970; Flores-Villela y Martínez-Salazar, 2009; Peterson et al., 1999). Por lo tanto, el agrupamiento de los haplotipos desde el sur del río Santo Domingo hasta las Tierras Altas de Chiapas fue inesperado (i.e., grupo SUR). Sin embargo, las relaciones halladas en la red de haplotipos y en la inferencia bayesiana mostraron a los haplotipos, al este del istmo, como un grupo notablemente más distanciado del resto del grupo SUR, lo que evidencia que esos haplotipos son únicos.
Este efecto de barrera intermitente del istmo de Tehuantepec ha sido observado en otros vertebrados (Barber y Klicka, 2010; Hardy et al., 2013; Sullivan et al., 1997, 2000) y en plantas (Ornelas et al., 2010). Los pasos mutacionales hallados en los análisis sugieren que los haplotipos al este del istmo de Tehuantepec divergieron del grupo SUR en un tiempo relativamente similar a la divergencia entre los grupos CENTRO y SUR. Hasta la fecha, se sabe poco sobre la posible conectividad de los bosques de niebla a través del istmo de Tehuantepec en los ciclos glaciales-interglaciales ocurridos a través de los últimos 400,000 años (Campbell, 1984). El registro de un espécimen de musaraña identificado como C. mexicanus (Choate, 1970) en una zona montañosa en medio del istmo de Tehuantepec (California Academy of Sciences, Mammalogy Collection, 14636), sugiere una distribución continua de las musarañas a través de un puente de bosque en el pasado. Un mayor tamaño de muestra de C. mexicanus en esta región del istmo, así como de las Tierras Altas de Chiapas, podría ayudar a esclarecer la posible conectividad de especies del bosque de niebla a través del Cuaternario (Mendelson III y Campbell, 1999; Hardy et al., 2013).
Finalmente, las tierras bajas de la provincia Veracruzana separan a la Sierra de Los Tuxtlas de la Sierra Madre Oriental y de la Sierra Madre del Sur. Con base en la divergencia genética observada en C. nelsoni (> 5 %, LOS TUXTLAS), esta especie se ha mantenido en aislamiento geográfico, probablemente durante al menos el Cuaternario tardío (Guevara, 2019; Woodman y Timm, 1999). Algo similar ha ocurrido con otros taxones endémicos de Los Tuxtlas (e.g., anfibios, reptiles y aves; Flores-Villela y Martínez-Salazar, 2009; Ornelas et al., 2013). Lo anterior destaca el papel de este sistema montañoso como un promotor de especiación por aislamiento geográfico (Chávez-Pesqueira y Núñez-Farfán, 2016; Ruiz-Sánchez y Ornelas, 2014), debido a la extensa llanura de tierras bajas a lo largo de la provincia Veracruzana, que potencialmente ha dificultado la dispersión de especies asociadas a ecosistemas de montaña.
Hasta la fecha, las evaluaciones taxonómicas de las especies del clado Cryptotis mexicanus, C. obscurus y C. nelsoni se han basado en caracteres morfológicos (Woodman y Timm, 1999), lo cual ha brindado un avance notable en su clasificación taxonómica. Sin embargo, debido a la relativa baja variación morfológica entre las especies, aún quedan dudas acerca del límite entre especies y la posible existencia de linajes crípticos (Guevara y Sánchez-Cordero, 2018a). Los patrones filogeográficos del gen Citb revelados en el presente estudio ofrecen una visión complementaria para esclarecer la distribución espacial de los linajes dentro de este grupo y asistir en la toma de decisiones taxonómicas.
Primero, el grupo de LOS TUXTLAS es un linaje independiente de los otros grupos cercanamente relacionados. La monofilia y los altos valores de apoyo, los pasos mutacionales y distancia genética con respecto al grupo más cercano, y el aislamiento geográfico, aportan evidencia concluyente para el reconocimiento específico de C. nelsoni (Guevara y Cervantes, 2014; He et al., 2015; Woodman y Timm, 1999), un taxón que previamente había sido considerado subespecie de C. mexicanus (Choate, 1970). Dentro del par de especies C. mexicanus y C. obscurus, el panorama es más complicado. Considerando los resultados de estudios previos y la evidencia que el gen Citb aporta en este estudio, consideramos necesaria una revisión taxonómica, en la que se podría evaluar la existencia de al menos 2 linajes evolutivos distribuidos desde el norte de la Sierra Madre Oriental hasta las Tierras Altas de Chiapas (Guevara y Sánchez-Cordero, 2018a; Ramírez-Pulido et al., 2004).
El primer linaje comprende al grupo NORTE, distribuyéndose desde la Reserva de la Biosfera El Cielo en la Sierra Madre Oriental, la Sierra de Otontepec al norte de la provincia Veracruzana y hasta el extremo oriental de la Faja Volcánica Transmexicana. Dado que las localidades tipo de C. mexicanus y C. obscurus están dentro del grupo NORTE y atendiendo al principio de prioridad, C. mexicanus sería el nombre válido para este linaje, y como consecuencia, C. obscurus sería un sinónimo junior de C. mexicanus. El segundo linaje comprendería a los grupos CENTRO y SUR, desde el sur del Pico de Orizaba hasta las Tierras Altas de Chiapas. Sin embargo, no se debe descartar la posibilidad de que los grupos CENTRO y SUR también representen unidades independientes, aunque el nivel de diferenciación no es de la misma magnitud que el observado entre los grupos LOS TUXTLAS, NORTE y SUR. Dentro de los grupos CENTRO y SUR, el río Santo Domingo marcaría el límite entre sus distribuciones. En cuanto al caso de la población de las Tierras Altas de Chiapas, este estudio genético no apoya el reconocimiento de su estado específico, tal como había sido sugerido anteriormente con base en evidencia morfológica (Guevara y Sánchez-Cordero, 2018a). En cualquier caso, es importante mencionar que no existen nombres disponibles para los linajes que abarcarían los grupos CENTRO y SUR. Un paso siguiente para resolver las incertidumbres taxonómicas deberá ser la inclusión de evidencia genética adicional, tal como genes nucleares, así como un mayor número de muestras provenientes del centro y sur de la Sierra Madre Oriental y al este del istmo de Tehuantepec. Además, es recomendable la evaluación de caracteres morfológicos del esqueleto poscraneal, los cuáles han sido útiles para la delimitación de especies del género.
Agradecimientos
A dos revisores anónimos por sus valiosos comentarios y observaciones. A F. A. Cervantes, L. León-Paniagua y C. Lorenzo por el apoyo para realizar el trabajo de campo y facilitar el préstamo de muestras para análisis genéticos. A C. Gómez-Trejo por su invaluable asistencia en el laboratorio de biología molecular, M. F. Ortiz-Ramírez por su ayuda en los análisis de inferencia bayesiana y R. M. Gámez-Pastrana por la revisión del manuscrito. FJ-V agradece al Programa de Movilidad de la Universidad Veracruzana por el apoyo para realizar su tesis de licenciatura. GH-C agradece a la U.S National Sience Fundation (NSF DEB-1754392, DEB-1441634) y al Conacyt (549963) por el apoyo financiero para una estancia postdoctoral. LG agradece al U.S National Sience Fundation (NSF 2002202) por el apoyo para trabajo de biología molecular.
Apéndice. Muestras utilizadas para análisis genéticos. Se incluye el nombre científico y la procedencia de las muestras, ya sea muestra de colección científica para la extracción de DNA o de la base de datos de secuencias genéticas GenBank.
Cryptotis mexicanus (CNMA 41946, 41949-41953, 46464-46465, 42724-42725, 42728, LGL*52-59; ECO-SC-M 7561, 7574; MZFC 8333, 8352-8353, 8368, 8376, 8385, 10370; 16220, 16222-16224; GenBank KT876861.1, 90959732, 90959734, 944547349, KF551836.1, KF551837.1, KF551838.1, KF551839.1, KT876861.1, MK286564.1, MK286565.1). C. obscurus (CNMA 44625-44627, 44631, 44637; GenBank KF551845.1, KF551846.1). C. nelsoni (CNMA 41962-41963, 41978, 41982-41984, 49290-49291, 49295-49296, 49298); GenBank KF551840.1, KF551841.1, KF551842.1, KF551843.1, KF551844.1). C. phillipsii (GenBank KF551850.1, KF551851.1, KF551852.1, KF551853.1). C. magnus (AB175139.1, AB175140.1, AB175141.1). C. alticola (KF551856.1). *LGL = catálogo personal de Lázaro Guevara (ejemplares en proceso de catalogación en la CNMA).
Referencias
Almendra, A. L., Rogers, D. S. y González-Cózatl, F. X. (2014). Molecular phylogenetics of the Handleyomys chapmani complex in Mesoamerica. Journal of Mammalogy, 95, 26–40. https://doi.org/10.1644/13-mamm-a-044.1
Anisimova, M. y Gascuel, O. (2006). Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Systematic Biology, 55, 539–552. https://doi.org/10.1080/10635150600755453
Barber, B. R. y Klicka, J. (2010). Two pulses of diversification across the Isthmus of Tehuantepec in a montane Mexican bird fauna. Proceedings of the Royal Society B: Biological Sciences, 277, 2675–2681. https://doi.org/10.1098/rspb.2010.0343
Barrier, E., Velasquillo, L., Chávez, M. y Gaulon, R. (1998). Neotectonic evolution of the Isthmus of Tehuantepec (southeastern Mexico). Tectonophysics, 287, 77–96. https://doi.org/10.1016/S0040-1951(98)80062-0
Burgin, C. J., Colella, J. P., Kahn, P. L. y Upham, N. S. (2018). How many species of mammals are there? Journal of Mammalogy, 99, 1-14. https://doi.org/10.1093/jmammal/gyx147
Caballero, M., Lozano-García, S., Vázquez Selem, L. y Ortega, B. (2010). Evidencias de cambio climático y ambiental en registros glaciales y en cuencas lacustres del centro de México durante el último máximo glacial. Boletín de la Sociedad Geológica Mexicana, 62, 359–377. https://doi.org/10.18268/BSGM2010v62n3a4
Campbell, J. A. (1984). A new species of Abronia (Sauria: Anguidae) with comments on the herpetogeography of the highlands of southern Mexico. Herpetologica, 40, 373–381.
Carleton, M. D., Sánchez, O. y Urbano-Vidales, G. (2002). A new species of Habromys (Muroidea: Neotominae) from México, with generic review of species definitions and remarks on diversity patterns among Mesoamerican small mammals restricted to humid montane forests. Proceedings of the Biological Society of Washington, 115, 488–533
Chávez-Pesqueira, M. y Núñez-Farfán, J. (2016). Genetic diversity and structure of wild populations of Carica papaya in Northern Mesoamerica inferred by nuclear microsatellites and chloroplast markers. Annals of Botany, 118, 1293-1306. https://doi.org/10.1093/aob/mcw183
Choate, J. R. (1970). Systematics and zoogeography of the Middle American shrews of the genus Cryptotis. University of Kansas publications, Museum of Natural History, 19, 195–317. https://doi.org/10.5962/bhl.part.15450
Churchfield, S. (1990). The natural history of shrews. Ithaca, NY: Cornell Univ. Press,
Excoffier, L. y Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10, 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Flores-Villela, O. y Martínez-Salazar, E. A. (2009). Historical explanation of the origin of the herpetofauna of Mexico. Revista Mexicana de Biodiversidad, 80, 817–833. https://doi.org/10.111/J.2007-8706
Fluxus Technology Ltd. (2015). Network 5.0.0.0 User Guide. December, 55.
Giarla, T. C. y Esselstyn, J. A. (2015). The challenges of resolving a rapid, recent radiation: empirical and simulated phylogenomics of Philippine shrews. Systematic Biology, 64, 727–740. https://doi.org/10.1093/sysbio/syv029
Guevara, L. (2017). They can dig it: semifossorial habits of the Mexican small-eared shrew (Mammalia: Cryptotis mexicanus). Revista Mexicana de Biodiversidad, 88, 1003–1005. https://doi.org/10.1016/j.rmb.2017.10.012
Guevara, L. (2019). Paleodistribution modelling for planning the growth of natural history collections. Revista Mexicana de Biodiversidad, 90, e902953. http://dx.doi.org/10.22201/ib.20078706e.2019.90.2953
Guevara, L. (2020). Altitudinal, latitudinal and longitudinal responses of cloud forest species to Quaternary glaciations in the northern Neotropics. Biological Journal of the Linnean Society, 130, 615–625. https://doi.org/10.1093/biolinnean/blaa070
Guevara, L. y Cervantes, F. A. (2014). Molecular systematics of small-eared shrews (Soricomorpha, Mammalia) within Cryptotis mexicanus species group from Mesoamerica. Acta Theriologica, 59, 233–242. https://doi.org/10.1007/s13364-013-0165-6
Guevara, L., Cervantes, F. A. y Sánchez-Cordero, V. (2015). Riqueza, distribución y conservación de los topos y las musarañas (Mammalia, Eulipotyphla) de México. Therya, 6, 43–68. https://doi.org/10.12933/therya-15-211
Guevara, L. y Sánchez-Cordero, V. (2018a). Patterns of morphological and ecological similarities of small-eared shrews (Soricidae, Cryptotis) in tropical montane cloud forests from Mesoamerica. Systematics and Biodiversity, 16, 551–564. https://doi.org/10.1080/14772000.2018.1470582
Guevara, L. y Sánchez-Cordero, V. (2018b). New records of a critically endangered shrew from Mexican cloud forests (Soricidae, Cryptotis nelsoni) and prospects for future field research. Biodiversity Data Journal, 6, e26667. https://doi.org/10.3897/BDJ.6.e26667
Guillot, G., Mortier, F. y Estoup, A. (2005). Geneland: a computer package for landscape genetics. Molecular Ecology Notes, 5, 712–715. https://doi.org/10.1111/j.1471-8286.2005.01031.x
Hardy, D. K., González-Cózatl, F. X., Arellano, E. y Rogers, D. S. (2013). Molecular phylogenetics and phylogeographic structure of Sumichrast’s harvest mouse (Reithrodontomys sumichrasti: Cricetidae) based on mitochondrial and nuclear DNA sequences. Molecular Phylogenetics and Evolution, 68, 282–292. https://doi.org/10.1016/j.ympev.2013.03.028
He, K., Woodman, N., Boaglio, S., Roberts, M., Supekar, S. y Maldonado, J. E. (2015). Molecular phylogeny supports repeated adaptation to burrowing within small-eared shrews genus of Cryptotis (Eulipotyphla, Soricidae). Plos One, 10, 1–13. https://doi.org/10.1371/journal.pone.0140280
Hope, A. G., Stephens, R. B., Mueller, S. D., Tkach, V. V. y Demboski, J. R. (2020). Speciation of North American pygmy shrews (Eulipotyphla: Soricidae) supports spatial but not temporal congruence of diversification among boreal species. Biological Journal of the Linnean Society, 129, 41–60. https://doi.org/10.1093/biolinnean/blz139
Irwin, D. M., Kocher, T. D. y Wilson, A. C. (1991). Evolution of the cytochrome b gene of mammals. Journal of Molecular Evolution, 32, 128–144. https://doi.org/10.1007/BF02515385
Jacquet, F., Hutterer, R., Nicolas, V., Decher, J., Colyn, M., Couloux, A. et al. (2013). New status for two African giant forest shrews, Crocidura goliath goliath and C. goliath nimbasilvanus (Mammalia: Soricomorpha), based on molecular and geometic morphometric analyses. African Zoology, 48, 13–29. https://doi.org/10.1080/15627020.2013.11407565
Kumar, S., Stecher, G., Li, M., Knyaz, C. y Tamura, K. (2018). Mega X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. https://doi.org/10.1093/molbev/msy096
Lefort, V., Longueville, J. E. y Gascuel, O. (2017). SMS: Smart Model Selection in PhyML. Molecular Biology and Evolution, 34, 2422–2424. https://doi.org/10.1093/molbev/msx149
Librado, P. y Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Lorenzo, C., Bolaños-Citalán, J., Navarrete-Gutiérrez, D., Pérez-López, J. A. y Guevara, L. (2019). In search of shrews of Chiapas: analysis of their distribution and conservation. Therya, 10, 121-129. https://doi.org/10.12933/therya-19-717
Mayen-Zaragoza, M., Guevara, L., Hernández-Canchola, G. y León-Paniagua, L. (2019). First record of shrews (Eulipotyphla, Soricidae) in the Sierra de Otontepec, an isolated mountain in Veracruz, Mexico. Therya, 10, 59–64. https://doi.org/10.12933/therya-19-690
Mendelson III, J. R. y Campbell, J. A. (1999). The taxonomic status of populations referred to Hyla chaneque in southern Mexico, with the description of a new treefrog from Oaxaca. Journal of Herpetology, 33, 80–86. https://doi.org/10.2307/1565545
Noguera-Urbano, E. A., Colmenares-Pinzón, J. E., Villota, J., Rodríguez-Bolaños, A. y Ramírez-Chaves, H. E. (2019). The shrews (Cryptotis) of Colombia: What do we know about them? Therya, 10, 131–147. https://doi.org/10.12933/therya-19-760
Ornelas, J. F., Ruiz-Sánchez, E. y Sosa, V. (2010). Phylogeography of Podocarpus matudae (Podocarpaceae): Pre-Quaternary relicts in northern Mesoamerican cloud forests. Journal of Biogeography, 37, 2384–2396. https://doi.org/10.1111/j.1365-2699.2010.02372.x
Ornelas, J. F., Sosa, V., Soltis, D. E., Daza, J. M., González, C., Soltis, P. S. et al. (2013). Comparative phylogeographic analyses illustrate the complex evolutionary history of threatened cloud forests of northern Mesoamerica. Plos One, 8, e56283. https://doi.org/10.1371/journal.pone.0056283
Peterson, A. T., Soberón, J. y Sánchez-Cordero, V. (1999). Conservatism of ecological niches in evolutionary time. Science, 285, 1265–1267. https://doi.org/10.1126/science.285.5431.1265
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. y Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67, 901–904. https://doi.org/10.1093/sysbio/syy032
Ramírez-Barahona, S. y Eguiarte, L. E. (2013). The role of glacial cycles in promoting genetic diversity in the Neotropics: the case of cloud forests during the Last Glacial Maximum. Ecology and Evolution, 3, 725–738. https://doi.org/10.1002/ece3.483
Ramírez-Pulido, J., Castillo-Morales, A., Salame-Méndez, A. y Castro-Campillo, A. (2004). Características morfológicas y morfométricas de cinco especies de Cryptotis (Mammalia: Soricomorpha). Acta Zoológica Mexicana, 20, 9–37. https://doi.org/10.21829/azm.2004.2022325
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S. et al. (2012). Mrbayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61, 539–542. https://doi.org/10.1093/sysbio/sys029
Ruiz-Sanchez, E. y Ornelas, J. F. (2014). Phylogeography of Liquidambar styraciflua (Altingiaceae) in Mesoamerica: survivors of a Neogene widespread temperate forest (or cloud forest) in North America? Ecology and Evolution, 4, 311–328. https://doi.org/10.1002/ece3.938
Smith, M. F. y Patton, J. L. (1999). Phylogenetic relationships and the radiation of sigmodontine rodents in South America: evidence from cytochrome b. Journal of Mammalian Evolution, 6, 89–128.
Starcová, M., Vohralík, V., Kryštufek, B., Černá Bolfíková, B. y Hulva, P. (2016). Phylogeography of the alpine shrew, Sorex alpinus (Soricidae, Mammalia). Folia Zoologica, 65, 107–116. https://doi.org/10.25225/fozo.v65.i2.a6.2016
Sullivan, J., Arellano, E. y Rogers, D. S. (2000). Comparative phylogeography of Mesoamerican highland rodents: Concerted versus independent response to past climatic fluctuations. American Naturalist, 155, 755–768. https://doi.org/10.1086/303362
Sullivan, J., Markert, J. A. y Kilpatrick, C. W. (1997). Phylogeography and molecular systematics of the Peromyscus aztecus species group (Rodentia: Muridae) inferred using parsimony and likelihood. Systematic Biology, 46, 426–440. https://doi.org/10.2307/2413690
Vallejo, R. M. y González-Cózatl, F. X. (2012). Phylogenetic affinities and species limits within the genus Megadontomys (Rodentia: Cricetidae) based on mitochondrial sequence data. Journal of Zoological Systematics and Evolutionary Research, 50, 67–75. https://doi.org/10.1111/j.1439-0469.2011.00634.x
Vega, R., McDevitt, A. D., Stojak, J., Mishta, A., Wójcik, J. M., Kryštufek, B. et al. (2020). Phylogeographical structure of the pygmy shrew: revisiting the roles of southern and northern refugia in Europe. Biological Journal of the Linnean Society, 129, 901–917. https://doi.org/10.1093/biolinnean/blz209
Vuilleumier, S. y Fontanillas, P. (2007). Landscape structure affects dispersal in the greater white-toothed shrew: inference between genetic and simulated ecological distances. Ecological Modelling, 201, 369–376. https://doi.org/10.1016/j.ecolmodel.2006.10.002
Woodman, N. (2011). Patterns of morphological variation amongst semifossorial shrews in the highlands of Guatemala, with the description of a new species (Mammalia, Soricomorpha, Soricidae). Zoological Journal of the Linnean Society, 163, 1267–1288. https://doi.org/10.1111/j.1096-3642.2011.00754.x
Woodman, N. y Timm, R. M. (1999). Geographic variation and evolutionary relationships among broad-clawed shrews of the Cryptotis goldmani group. Fieldiana, Zoology, 91, 1–35.
Woodman, N., Matson, J. O., McCarthy, T. J., Eckerlin, R. P., Bulmer, W. y Ordóñez-Garza, N. (2012). Distributional records of shrews (Mammalia, Soricomorpha, Soricidae) from Northern Central America with the first record of Sorex from Honduras. Annals of Carnegie Museum, 80, 207–237.


