
Víctor Julio Piñeros a, b y Nancy Calderón-Cortés a, *
a Universidad Nacional Autónoma de México, Escuela Nacional de Estudios Superiores, Unidad Morelia, Laboratorio de Ecología Molecular, Antigua carretera a Pátzcuaro 8701, Ex-Hacienda de San José de la Huerta, 58190 Morelia, Michoacán, México
b Universidad de Guadalajara, Centro Universitario de la Costa Sur, Departamento de Estudios para el Desarrollo Sustentable de Zonas Costeras, Gómez Farías 82, 48980 San Patricio-Melaque, Jalisco, México
*Autor para correspondencia: ncalderon@enesmorelia.unam.mx (N. Calderón-Cortés)
Recibido: 19 noviembre 2021; aceptado: 17 enero 2023
Resumen
El papel de los peces en la dinámica trófica de los ecosistemas acuáticos se ha estudiado usando diferentes métodos que permiten determinar su dieta y posición trófica (inspección visual del contenido estomacal y heces, isótopos estables, secuenciación de ADN). Con los avances tecnológicos en la secuenciación de alto rendimiento del ADN, el uso del método de metabarcoding se ha incrementado en los últimos años, demostrando tener mayor precisión y alcance taxonómico que otros métodos, además de que permite el análisis simultáneo de un mayor número de muestras en un menor tiempo. En esta revisión, se describen los pasos del método de metabarcoding, discutiendo sus ventajas y limitaciones, así como las alternativas experimentales y analíticas propuestas para atender dichas limitaciones. Además, se presenta una síntesis del estado del conocimiento del método de metabarcoding aplicado al análisis de la ecología trófica de peces para entender los alcances y las limitaciones de este método en México.
Palabras clave: Biodiversidad; Código de barras; Dietas; Genes; Nicho trófico
© 2023 Universidad Nacional Autónoma de México, Instituto de Biología. Este es un artículo Open Access bajo la licencia CC BY-NC-ND
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Metabarcoding: a promising tool for studying the trophic ecology of Mexican fish
Abstract
The role of fishes in trophic dynamics in the aquatic ecosystems has been studied by analyzing fish diet and trophic position in ecosystems using diverse methods (e.g., visual inspection of stomach contents and feces, stable isotopes, DNA sequencing). Thanks to the advances in high-throughput DNA sequencing technology, the use of the metabarcoding method has increased in recent years, proving to have greater precision and taxonomic scope than other methods, and allowing the analysis of a larger number of samples simultaneously in less time. This review describes the metabarcoding method, discusses its advantages and limitations, and presents experimental and analytical alternatives to address such limitations. Additionally, we provide a synthesis of the current state of knowledge of the metabarcoding method applied to the analysis of the trophic fish ecology to understand the scope and limitations of using this method in Mexico.
Keywords: Biodiversity; Barcoding; Diets; Genes; Trophic niche
© 2023 Universidad Nacional Autónoma de México, Instituto de Biología. This is an open access article under the CC BY-NC-ND license
Introducción
Desde el punto de vista funcional, las relaciones alimenticias de los organismos en los ecosistemas representan un componente importante de la ecología trófica, la cual tiene como objetivo determinar la funcionalidad ecológica de las especies a través del papel trófico que cumplen dentro de su comunidad biológica, el nivel de redundancia ecológica de las especies, el flujo de energía y la capacidad de resiliencia de los ecosistemas (Cartes et al., 2016; Siegenthaler et al., 2019). El papel trófico de una especie en una red alimenticia se determina por su nivel trófico, lo que come (información cualitativa) y en qué proporción (información cuantitativa), es decir su dieta (da Silveira et al., 2019; Finlay et al., 2010). La dieta de un organismo puede determinarse mediante: el análisis visual del contenido estomacal, técnicas indirectas como el uso de biomarcadores e isótopos estables y técnicas basadas en ADN que permiten la identificación molecular de los organismos presentes en el contenido estomacal o heces fecales (da Silveira et al., 2019). Los diferentes métodos de análisis de dieta han mejorado continuamente y se han actualizado en atención a las limitaciones técnicas (da Silveira et al., 2019). Sin embargo, debido a los avances en la tecnología para la secuenciación del ADN, el uso de la secuenciación de alto rendimiento (HTS por sus siglas en inglés) o secuenciación masiva para el estudio de la dieta mediante la caracterización genética o “metabarcoding”, ha incrementado notablemente en los últimos años (Alberdi et al., 2019; Arranz et al., 2020; Deagle et al., 2019; Ducotterd et al., 2021).
El “metabarcoding” ha surgido como una herramienta metodológica útil para la identificación de unidades taxonómicas dentro de una muestra, permitiendo el estudio de la diversidad de comunidades biológicas donde el muestreo es difícil y el monitoreo a largo plazo es importante dado el deterioro progresivo de los ecosistemas en los últimos años (Arranz et al., 2020; Leray et al., 2013). Este método se basa en la secuenciación de alto rendimiento de marcadores de ADN en muestras de contenido estomacal o heces fecales (i.e., muestras ambientales) y la identificación de múltiples taxones presentes en el ADN total extraído de la muestra (Arranz et al., 2020; Siegenthaler et al., 2019; Tournayre et al., 2020). La estrategia de análisis de metabarcoding se ha usado para determinar la composición de la dieta de un amplio rango de animales incluyendo mamíferos, aves, reptiles, moluscos, artrópodos y peces (Brandl et al., 2020; Deagle et al., 2019; Ducotterd et al., 2021); para mejorar la precisión en la estimación de dietas complejas presentes en especies omnívoras y generalistas (Duccotterd et al., 2021); para probar hipótesis sobre selección y/o preferencias de presas (Vesterinen et al., 2016); y para la reconstrucción de redes de interacciones ecológicas (Cuff et al., 2022).
El metabarcoding en general es considerado un método sencillo, rápido y relativamente accesible que tiene el potencial de mejorar la precisión en el análisis de la dieta y el estudio de la ecología trófica de los organismos estudiados, particularmente de especies cuyos patrones de alimentación son difíciles de observar (Alberdi et al., 2019; Ducotterd et al., 2021; Leray et al., 2015; Siegenthaler et al., 2019). A partir de una revisión reciente de 150 estudios que incluyen aproximadamente 250 especies, donde se comparó el uso del metabarcoding en el estudio trófico de organismos terrestres vs. organismos acuáticos, de Sousa et al. (2019) reportaron que los análisis de metabarcoding de organismos acuáticos demostraron mayor diversidad de presas en las dietas y por tanto mayor diversidad de interacciones tróficas, que inclusive han reposicionado el nivel trófico que previamente se había documentado para varias especies. Sin embargo, los estudios realizados con organismos acuáticos representan un menor porcentaje (< 50%) y dentro de este grupo, los peces son poco estudiados a pesar de su importancia en la ecología y salud de los ecosistemas acuáticos. Una revisión más reciente sobre el uso de las técnicas basadas en el análisis de ADN (PCR diagnóstico, PCR cuantitativo y metabarcoding), confirmó su potencial en el análisis trófico de las especies, al ofrecer una alta especificidad y sensibilidad en la detección de los ítems alimenticios, la capacidad de estandarizar la metodología, y la posibilidad de usar técnicas de secuenciación de alto rendimiento que permite procesar un gran número de muestras, obteniendo información confiable y cuantificable (Traugott et al., 2021). Sin embargo, en esta revisión solo se discuten 3 estudios de caso de metabarcoding de peces (Guillerault et al., 2017; Harms-Tuohy et al., 2016; Jakubaviciute et al., 2017), por lo que es difícil conocer el impacto real de esta herramienta en el análisis trófico de peces. La evaluación del potencial de aplicación de metodologías que permitan el estudio de la ecología trófica de peces en ambientes naturales es crucial, no solo para avanzar en el entendimiento de la dinámica trófica en los ecosistemas, sino para apoyar el desarrollo de estrategias de conservación y manejo sostenible de especies clave para el funcionamiento de los ecosistemas acuáticos (i.e., los peces loros en los ecosistemas arrecifales; Castro-Sanguino y Sánchez [2012]; los tiburones como depredadores superiores que regulan las comunidades marinas, Stevens et al. [2000], por mencionar algunos), y de especies de importancia comercial cuya ausencia o sobreexplotación podrían afectar la dinámica trófica de las comunidades biológicas (Link, 2002).
El objetivo de esta revisión es generar una síntesis sobre el uso del metabarcoding para el análisis de la dieta y la ecología trófica de peces, analizando las limitaciones tanto biológicas como técnicas que existen para su implementación, así como las alternativas experimentales y analíticas propuestas para disminuir su efecto. Además, se presenta un análisis de los estudios de dieta de peces basados en metabarcoding y se discute el potencial y factibilidad de esta estrategia metodológica para el estudio de la ecología trófica de peces de México.
El método del metabarcoding
El metabarcoding es una técnica basada en la amplificación por PCR de marcadores genéticos presentes en una gran variedad de especies, seguido por la secuenciación de regiones genéticas homólogas. Posteriormente, las secuencias obtenidas son comparadas con bibliotecas de referencia para la identificación taxonómica (Leray et al., 2013). A nivel tecnológico, el avance más significativo para este método ha sido el desarrollo de HTS, que generan un gran número (de miles a millones) de secuencias (códigos de barras) de ADN de diagnóstico a partir de muestras heterogéneas y/o muestras ambientales. Los beneficios del análisis de secuencias de ADN para la caracterización de la dieta y la identificación de presas en las muestras incluyen: la detección de organismos que no pueden ser identificados por métodos visuales (generalmente organismos de cuerpo blando que son digeridos con mayor rapidez), menor dependencia directa de la experiencia taxonómica, capacidad para detectar organismos que carecen de características morfológicas de diagnóstico, el procesamiento de varios cientos de muestras de manera simultánea, y la velocidad de análisis e idoneidad para procesos automatizados (Alberdi et al., 2019; Berry et al., 2015; Corse et al., 2019; Tournayre et al., 2020).
El flujo de trabajo general para los estudios de dieta que usan el método de metabarcoding (fig. 1) comienza con la recolecta y procesamiento de muestras en campo, seguido de la fase de laboratorio, en la cual es necesario extraer el ADN total de la muestra dietética con la mayor integridad posible (i.e., sin degradación) y libre de compuestos contaminantes que interfieran con su análisis (i.e., compuestos polifenólicos, ácidos húmicos; Juen y Traugott [2006] y Schrader et al. [2012]), y amplificar por PCR los marcadores de códigos de barras de ADN de los grupos taxonómicos alimentarios de interés con cebadores (“primers”) u oligonucleótidos específicos (fragmentos cortos de ADN complementarios a la secuencia del marcador genético que desea amplificar), para los cuales se haya probado previamente su eficiencia. Posteriormente, es necesario realizar la construcción de librerías para la secuenciación de los marcadores genéticos de ADN y para la clasificación taxonómica de las secuencias recuperadas. Una de las plataformas de HTS de mayor uso es Illumina (i.e., MiSeq y NextSeq). Para la secuenciación por Illumina, que genera decenas de millones a billones de fragmentos amplificados por muestra, es necesario ligar mediante PCR los adaptadores e índices de nucleótidos a los marcadores genéticos, lo cual permitirá identificar las bibliotecas y muestras de diferentes organismos (Alberdi et al., 2019; Meyer y Kischer, 2010; Miya et al., 2020). Una vez obtenidas las lecturas de la secuenciación, es necesario realizar un procesamiento de datos para la eliminación de los adaptadores e índices, así como la formación de secuencias quiméricas y secuencias de baja calidad, para finalmente, mediante el uso de herramientas y programas bioinformáticos, obtener una lista taxonómica de los organismos presentes en cada una de las muestras analizadas. A partir de esta lista taxonómica, que incluye el número de secuencias detectada para cada organismo identificado, es posible entonces realizar el análisis de la dieta (Alberdi et al., 2019; Miya et al., 2020). Si bien este flujo de trabajo se ha aplicado en una variedad de animales, cada uno de los pasos de este proceso tiene sus propias limitaciones y sesgos, y debe implementarse con cuidado. A continuación, se describen los principales sesgos y recomendaciones en cada uno de los pasos.
Diseño del estudio
Para realizar los estudios de metabarcoding se deben tener en cuenta detalles metodológicos relacionados con la biología e historia de vida de la especie de interés, como el área de distribución, tipo de hábitat, estacionalidad, horas de actividad, estatus de conservación, entre otros. Esta información determina la mejor técnica para la toma de muestra: una muestra invasiva a partir de la disección del tracto digestivo del organismo, o una muestra no invasiva como la recolecta de heces fecales o reflujo de contenido estomacal. El tipo de muestra además determina su preservación (etanol, desecación, dimetilsulfóxido, etc.); las recomendaciones específicas para la toma y preservación de muestras son revisadas con detalle por Alberdi et al. (2019), Creer et al. (2016) y Dickie et al. (2018). Independientemente del método de preservación de muestras empleado, es recomendable que todas las muestras se conserven y almacenen en condiciones idénticas para minimizar los sesgos técnicos específicos de la muestra (Alberdi et al., 2019).
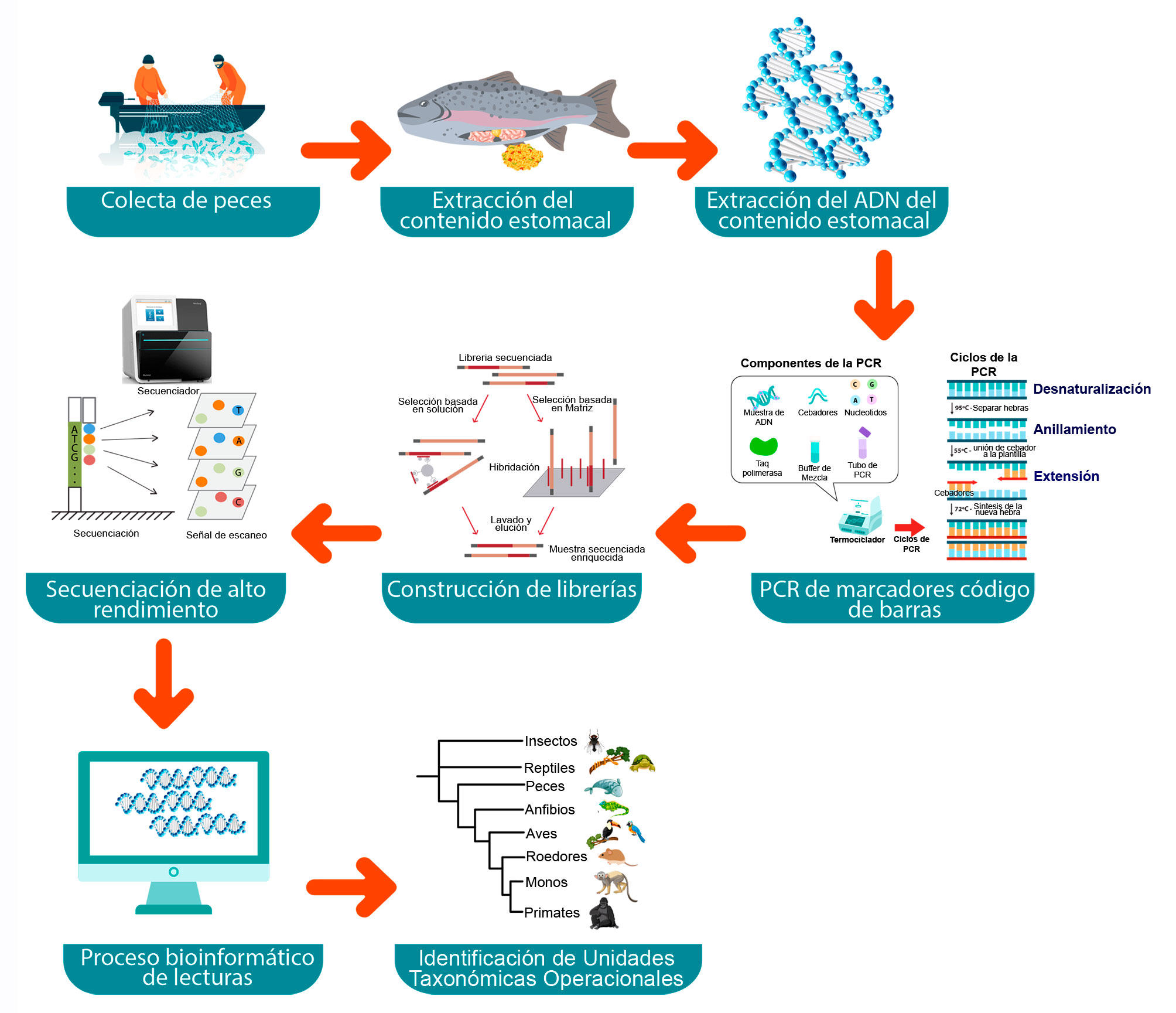
La amplificación del ADN es un paso crítico para el éxito de la técnica de metabarcoding, por lo que se requiere la selección de marcadores de ADN (código de barras) y de los oligonucleótidos cebadores apropiados para la identificación de los diferentes ítems alimenticios provenientes de una amplia comunidad biológica, por lo que a priori se necesita plantear un enfoque taxonómico para el análisis (Alberdi et al., 2019; da Silveira et al., 2019; Deagle et al., 2014). Esto determinará tanto la amplitud filogenética de los elementos dietéticos recuperables (i.e., peces, insectos, artrópodos, corales, plantas, hongos, etc.), como la resolución taxonómica que se logrará alcanzar (orden, familia, género o especie) (Elbrecht et al., 2019; Taberlet et al., 2012). Es importante asegurarse de que existan marcadores genéticos universales para los principales taxones presentes en la dieta que se desea analizar (Piñol et al., 2019), que el fragmento de ADN amplificado por los cebadores sea lo suficientemente variable para discriminar especies cercanas (CBOL, 2009; Hebert et al., 2003) y preferentemente corto (170-440 pb) para asegurar la amplificación usando ADN parcialmente degradado, lo cual es común en estudios de metabarcoding (Berry et al., 2015; Deagle et al., 2006, Leray et al., 2013), así como seleccionar los marcadores que cuenten con secuencias públicas en las bases de datos para una identificación taxonómica exitosa (Devloo-Delva et al., 2018; Elbrecht et al., 2016; Freeland et al., 2017; Tournayre et al., 2020). En la tabla 1 se incluye una lista de los principales marcadores genéticos de códigos de barras (y los oligonucleótidos cebadores correspondientes) utilizados para la identificación taxonómica de diferentes grupos biológicos.
Un aspecto importante que tiene que contemplarse en el diseño de los estudios de metabarcoding se refiere al control de falsos positivos (error tipo I) y falsos negativos (error tipo II), los cuales son comunes en este tipo de estudios (Alberdi et al., 2019; Corse et al., 2017; Piñol et al., 2014; Taberlet et al., 2018; Tounayre et al., 2020). Los falsos positivos corresponden a artefactos experimentales/moleculares (e.g., errores de PCR, cambio de etiquetas, contaminación ambiental, o contaminación cruzada entre muestras) que conducen a la detección de taxones que inicialmente no estaban presentes en la muestra (Corse et al., 2019). Por lo que este tipo de errores pueden controlarse a través de: i) un manejo cuidadoso de muestras (tanto en campo como en laboratorio, mediante la esterilización del material y separación de áreas pre y post-PCR); ii) la inclusión de réplicas experimentales en cada una de las técnicas usadas (extracción de ADN, PCR, etc.); iii) el uso de controles negativos, es decir, muestras que carecen de ADN; y iv) la inclusión de controles bioinformáticos, en los cuales se determine un número o un porcentaje mínimo de copias de cada secuencia como valor umbral para que los datos sean tomados en cuenta en los análisis posteriores (Corse et al., 2019; Cuff et al., 2022; Galan et al., 2018; Tournayre et al., 2020). Por su parte, los falsos negativos corresponden a taxones que no se detectan aun cuando están presentes en la muestra, los cuales comúnmente representan taxones raros, o taxones que presentan una baja concentración de ADN en la muestra, aunque también pueden estar asociados a la eficiencia en la amplificación del ADN (Alberdi et al., 2019; Corse et al., 2019; Cuff et al., 2022; Elbrecht y Leese, 2017). Para minimizar este tipo de errores, se recomienda usar varios marcadores moleculares de diferentes loci o varios cebadores para un locus que mejoren la precisión en la detección de la riqueza taxonómica presente en una muestra (Corse et al., 2019; Cuff et al., 2022; Tournayre et al., 2020).
Trabajo experimental
Las muestras utilizadas en los análisis de dieta son principalmente muestras fecales y contenidos estomacales, lo que implica trabajar con muestras de ADN degradado, en las cuales el método de extracción de ADN utilizado tiene un impacto en la cantidad y calidad del ADN extraído, repercutiendo posteriormente en la amplificación de los marcadores genéticos (Jedlicka et al., 2013; Majaneva et al., 2018; Oehm et al., 2011). Existen varios métodos utilizados en la extracción de ADN, especialmente con organismos metazoos, que se basan en el uso de una alta concentración de sales o de solventes orgánicos como el fenol-cloroformo (Sambrook et al., 1989). Aunque estos métodos son económicos y fáciles de seguir, utilizan sustancias altamente tóxicas que son difíciles de desechar. Otros métodos de extracción de ADN se realizan con sistemas comerciales con soluciones estandarizadas (e.g., Qiagen, QIAmp, ADN Stool Mini Kit y PowerFecal / Soil ADN Kit, o Zymo XpeditionTM Soil / Fecal ADN MiniPrep kit). Sin embargo, estos métodos son más costosos que los primeros y no siempre son efectivos debido a la naturaleza o composición química de la muestra. Por ejemplo, la presencia de polisacáridos y compuestos polifenólicos, la acidez de la muestra, entre otros, pueden inhibir los procesos técnicos en pasos posteriores del protocolo de metabarcoding, como la ligación de oligonucleótidos o la amplificación por PCR (Pandey et al., 1996). Por esta razón, se recomienda la estandarización de una técnica de extracción de ADN que incluya procedimientos que remuevan compuestos químicos contaminantes presentes en las muestras de interés, como la inclusión de un paso de purificación del ADN utilizando filtros que contienen sílice, los cuales han mostrado ser efectivos en muestras de contenido intestinal de escarabajos (Juen y Traugott, 2006). Otro punto a tener en cuenta es que las características tisulares de las presas en la dieta también pueden afectar la calidad y cantidad del ADN purificado, ya que la extracción de ADN de animales con estructuras esclerotizadas o calcáreas (insectos, zooplancton, moluscos) es más difícil que las de especies de cuerpo blando. Además, la proporción de ADN de las presas puede variar en la muestra, ya que éstas pueden presentar diferentes tasas de digestión en los intestinos del consumidor (i.e., por diferencias en condiciones ambientales como temperatura), ser afectadas de forma variable por el metabolismo del consumidor, por el tiempo de ingestión y por el volumen de tejido consumido, repercutiendo en la capacidad de detectar ciertos taxones dentro de la muestra, particularmente en especies generalistas, las cuales pueden consumir un amplio rango de especies, algunas en muy baja frecuencia (Cuff et al., 2022; Traugott et al., 2021). Para lidiar con este problema se ha propuesto la realización de experimentos de calibración alimenticia, que permiten entender los procesos digestivos y el tiempo de transición intestinal de las presas que constituyen la dieta, estimando su tasa de digestión (Traugott et al., 2021). Sin embargo, determinar el tiempo de digestión de diferentes presas en la dieta de un pez, representa un reto metodológico. Algunos experimentos de este tipo realizados con peces, han demostrado que la detección de ADN de huevos o larvas de peces empieza a disminuir aproximadamente 2 horas después de la ingestión, con un rango máximo de detección de ente 24 y 48 horas (Hunter et al., 2012; Traugott et al., 2021).
Tabla 1
A) Lista de oligonucleótidos cebadores universales de marcadores de códigos de barras para la identificación taxonómica y construcción de colecciones moleculares de varios grupos taxonómicos: metazoos (invertebrados y vertebrados), procariotas, hongos y plantas. B) Lista de cebadores para marcadores metabarcoding universales diseñados y probados en metazoos acuáticos. C) Lista de oligonucleótidos cebadores degenerados desarrollados para la identificación taxonómica de diferentes grupos.
| Grupo taxonómico | Marcador molecular | Nombre de cebadores | Cebadores | Longitud del amplificado (pb) | Muestra biológica | Referencia |
| A) Cebadores universales para marcadores códigos de barras por secuenciación Sanger para enriquecimiento de bases de datos de referencia | ||||||
| Metazoo | COI | LCO1490 | GGTCAACAAATCATAAAGATATTGG | 710 | Identificación taxonómica y construcción de colecciones moleculares | Folmer et al., 1994 |
| HC02198 | TAAACTTCAGGGTGACCAAAAAATCA | |||||
| Peces | FishF1 | TCAACCAACCACAAAGACATTGGCAC | 655 | Ward et al., 2005 | ||
| Peces | FishR1 | TAGACTTCTGGGTGGCCAAAGAATCA | ||||
| Metazoo | Cytb | mcb398 | TACCATGAGGACAAATATCATTCTG | 421 | Verma y Singh, 2003 | |
| mcb869 | CCTCCTAGTTTGTTAGGGATTGATCG | |||||
| Metazoo | 16S | 16s SAR-L | CGCCTGTTTATCAAAAACAT | 511-571 | Palumbi, 1996 | |
| 16s SBR_H | CCGGTCTGAACTCAGATCACGT | |||||
| Procariotas | 16S | 341F | GGTDTTACCGCGGCKGCTG | 180 | Klindworth et al., 2013 | |
| 518R | GGTDTTACCGCGGCKGCTG | |||||
| Eucariotas | 18S | 1389F | TCCCTGCCHTTTGTACACAC | 130 | Malviya et al., 2016 | |
| 1510R | CCTTCYGCAGGTTCACCTAC | |||||
| Metazoo | 12S | L1091 | AAAAAGCTTCAAACTGGGATTAGATACCCCACTAT | 386 | Kocher et al., 1989 | |
| H1478 | TGACTGCAGAGGGTGACGGGCGGTGTGT | |||||
| Hongos | ITS | ITS1 | TCCGTAGGTGAACCTGCGG | 290 | White et al., 1990 | |
| ITS5 | GGAAGTAAAAGTCGTAACAAGG | 315 | ||||
| ITS2 | GCTGCGTTCTTCATCGATGC | 290 | ||||
| ITS3 | GCATCGATGAAGAACGCAGC | 330 | ||||
| ITS4 | TCCTCCGCTTATTGATATGC | |||||
| Plantas | rbcL | rbcL724R | TCGCATGTACCTGCAGTAGC | 470-588 | Ivanova et al., 2008 | |
| rbcL1F | ATGTCACCACAAACAGAGACTAAAGC | |||||
| Plantas | matK | matK472F | CCCRTYCATCTGGAAATCTTGGTTC | 600-800 | Yu et al., 2011 | |
| matK1248R | GCTRTRATAATGAGAAAGATTTCTGC | |||||
| Plantas | trnH-psbA | trnH | CGCGCATGGTGGATTCACAATCC | 274-594 | Simpson et al., 2003 | |
| psbA | GTTATGCATGAACGTAATGCTC | Sang et al., 1997 | ||||
| Algas y cianobacterias | 23S | P23SrV_f1 | GGACAGAAAGACCCTATGAA | 377-414 | Sherwood y Presting, 2007 | |
| P23SrV_r1 | TCAGCCTGTTATCCCTAGAG | |||||
| B) Cebadores para el análisis de Metabarcoding | ||||||
| Metazoo | COI | mlCOIintF | GGWACWGGWTGAACWGTWTAYCCYCC | 315-319 | Contenido estomacal | Leray et al., 2013 |
| mlCOIintR | GGRGGRTASACSGTTCASCCSGTSCC | |||||
| Metazoo | coi.175f | GGAGGCTTTGGMAAYTGRYT | 55 | ADN ambiental | Collins et al., 2019 | |
| coi.226r | GGGGGAAGAARYCARAARCT | |||||
| Metazoo | coi.175f | GGAGGCTTTGGMAAYTGRYT | 130 | |||
| coi.345r | TAGAGGRGGGTARACWGTYCA | |||||
| Invertebrados | LepF1 | CCATCTCATCCCTGCGTGT-CTCCGACTCAGNNNNNNNATTCHACDAAYCAYAA RGAYATYGG | 133 | Muestras fecales | Gillet et al., 2015 | |
| Tabla 1. Continúa | ||||||
| Grupo taxonómico | Marcador molecular | Nombre de cebadores | Cebadores | Longitud del amplificado (pb) | Muestra biológica | Referencia |
| Invertebrados | EPT-long-univR | CCTCTCTATGGGCAGTCGGTGATNNNNNNNACTATAAAARAAAATYTDAYAAA DGCRTG | ||||
| Peces | Cytb | L14912-CYB | TTCCTAGCCATACAYTAYAC | 235 | Muestras de agua | Minamoto et al., 2012 |
| Peces | H15149-CYB | GGTGGCKCCTCAGAAGGACATTTGKCCYCA | ||||
| Metazoo | 12S | MiFish-U-F | GTCGGTAAAACTCGTGCCAGC | 171 | Muestras de agua | Miya et al., 2015 |
| Metazoo | MiFish-U-R | CATAGTGGGGTATCTAATCCCAGTTTG | ||||
| Metazoo | MiFish-E-F | GTTGGTAAATCTCGTGCCAGC | ||||
| Metazoo | MiFish-E-R | CATAGTGGGGTATCTAATCCTAGTTTG | ||||
| Metazoo | Tele 02-f | AAACTCGTGCCAGCCACC | 167-171 | Taberlet et al., 2018 | ||
| Metazoo | Tele 02-r | GGGTATCTAATCCCAGTTTG | ||||
| Metazoo | Elas02-f | GTTGGTHAATCTCGTGCCAGC | ||||
| Metazoo | Elas02-r | CATAGTAGGGTATCTAATCCTAGTTTG | ||||
| Metazoo | L1848 | ACACCGCCCGTCACTCT | 63 | Validación in silico | Valentini et al., 2016 | |
| Metazoo | H1913 | CTTCCGGTACACTTACCATG | ||||
| Metazoo | 12S-V5f | ACTGGGATTAGATACCCC | 106 | Validación in silico y análisis de muestras fecales | Riaz et al., 2011 | |
| Metazoo | 12S-V5r | TAGAACAGGCTCCTCTAG | ||||
| Metazoo | 16S | Fish16sF/D | GACCCTATGGAGCTTTAGAC | 219 | Contenido estomacal | Berry et al., 2015 |
| Metazoo | 16s2R | CGCTGTTATCCCTADRGTAACT | ||||
| Metazoo | 16S-R1-F | GTCYHGCCTGCCCHGTGA | 440 | Contenido estomacal | Devloo-Delva et al., 2018 | |
| Metazoo | 16S-R2-R | CCAACATCGAGGTCGTAARCC | ||||
| Algas y cianobacterias | 23S | P23SrV_f1 | GGACAGAAAGACCCTATGAA | 377-414 | Contenido estomacal | Sherwood y Presting, 2007 |
| P23SrV_r1 | TCAGCCTGTTATCCCTAGAG | |||||
| C) Cebadores degenerados para estudios de Metabarcoding | ||||||
| Metazoo | COI | mlCOIintF-XT | GGWACWRGWTGRACWITITAYCCYCC | 313 | Muestras biológicas de litorales rocosos | Wangensteen et al., 2018 |
| Metazoo | mlCOIintF-XT* | GGWACWRGWTGRACWGTYTAYCCYCC | ||||
| Metazoo | jgHCO2198 | TAIACYTCIGGRTGICCRAARAAYCA | ||||
| Metazoo | jgHCO2198* | TAKACYTCWGGRTGRCCRAARAAYCA | ||||
| Macroinvertebrados | fwhF1 | YTCHACWAAYCAYAARGAYATYGG | 178-205 | Experimentos y validación in silico | Vamos et al., 2017 | |
| Macroinvertebrados | fwhR1 | ARTCARTTWCCRAAHCCHCC | ||||
| Macro-invertebrados | fwhF2 | GGDACWGGWTGAACWGTWTAYCCHCC | ||||
| Macro-invertebrados | fwhR2 | GTRATWGCHCCDGCAARWACWGG | ||||
| Macro-invertebrados | fwhR2n | GTRATWGCHCCDGCTARWACWGG | ||||
| Macro-invertebrados acuáticos | BF1 | ACWGGWTGRACWGTNTAYCC | 217-421 | Experimentos y validación in silico | Elbrecht y Lesse, 2017 | |
| Macro-invertebrados acuáticos | BF2 | GCHCCHGAYATRGCHTTYCC | ||||
| Macro-invertebrados acuáticos | BR1 | ARYATDGTRATDGCHCCDGC | ||||
| Macro-invertebrados acuáticos | BR2 | TCDGGRTGNCCRAARAAYCA |
Un inconveniente común en los análisis moleculares de muestras de contenido estomacal o heces fecales es la contaminación de ADN proveniente del individuo depredador. El ADN de las presas tiende a estar mucho más degradado que el ADN de los depredadores, por lo que la sobreabundancia del ADN amplificable del depredador aumenta con el tamaño del fragmento que se desea amplificar. Los métodos de PCR no son lo suficientemente sensibles para detectar secuencias con poca abundancia y utilizan cebadores universales que amplifican por igual, tanto el ADN del depredador como de las presas (Lui et al., 2019; Su et al., 2018; Vestheim y Jarman, 2008). Se han propuesto varias metodologías para lidiar con este problema como el filtrado bioinformático y exclusión de las secuencias del depredador (Cuff et al., 2022), pero el método más usado consiste en sintetizar cebadores específicos para bloquear el ADN del depredador en el PCR (blocking primers). En muchos casos, estos cebadores de bloqueo consisten en un espaciador de 3 hidrocarbonos (C) en el extremo 3’, lo cual detiene la reacción de elongación sin afectar las propiedades de unión del cebador (Lui et al., 2019; Su et al., 2018; Vestheim y Jarman, 2008). Esto previene la amplificación de ADN del depredador, sin perjudicar la amplificación del ADN de las presas en la dieta (Su et al., 2018). Una de las desventajas de utilizar esta técnica es que la presencia de canibalismo dentro de las especies de peces es común, y el uso de cebadores bloqueados impediría tener evidencia de este comportamiento biológico (Dahl et al., 2017; French et al., 2020; Waraniak et al., 2019). Además, en varios estudios se ha encontrado evidencia de contaminación por depredación secundaria. Esta contaminación se refiera a la detección de la dieta de la presa consumida que es detectada en la dieta del depredador (Berry et al., 2015; Bowser et al., 2013). Si bien esto puede ser considerado un problema, algunos autores lo han visto como la posibilidad de explorar con mayor profundidad la red trófica (Berry et al., 2015).
Análisis bioinformático
Los datos provenientes del secuenciador son lecturas crudas sin procesar que están fragmentadas, contienen errores, y/o se ven afectadas por profundidades de secuenciación desiguales (Ducotterd et al., 2021). Por lo tanto, para analizar con precisión estos datos, es necesario ensamblar las lecturas de secuencias obtenidas en segmentos contiguos más grandes llamados “contigs” utilizando programas computacionales especiales (Anantharaman et al., 2016). El uso de un ensamblador que produzca “contigs” largos permitirá una determinación taxonómica más precisa de los organismos (van der Walt et al., 2017). Por esta razón, se han desarrollado múltiples procedimientos bioinformáticos o “pipelines” para ensamblar secuencias sin procesar, simplemente fusionando lecturas de extremos emparejados o mediante ensamblado de novo (van der Walt et al., 2017). Sin embargo, existen diferentes alternativas tanto de ensambladores como de protocolos de ensamblado, por lo cual se necesitan métodos de control y validación que permitan parametrizar los procedimientos bioinformáticos. Esto se puede lograr creando y secuenciando comunidades simuladas también conocidas como “mock communities”, que son referencias para bases de datos de ADN y se utilizan como controles positivos para la secuenciación de alto rendimiento (Jusino et al., 2018).
El problema más importante del análisis bioinformático es la disponibilidad de secuencias de referencia para la asignación taxonómica de las secuencias ensambladas (Alberdi et al., 2019; Berry et al., 2015; Elbrecht et al., 2016). Esto se debe a que, así como la morfología no puede proporcionar identidades para especies no incluidas en las claves taxonómicas, la identificación estricta de las muestras por marcadores genéticos o códigos de barras de ADN no puede realizar asignaciones donde las colecciones de referencia están incompletas, o donde el marco taxonómico subyacente está poco estudiado.
La asignación de especies requiere una base de datos de referencia que contenga secuencias asignadas taxonómicamente. Existen diferentes bases de referencia generales, con diferentes niveles de curaduría taxonómica como el GenBank del NCBI y Barcode Of Life Database (BOLD), así como bases de datos específicas para algunos grupos taxonómicos como SILVA y UNITE (tabla 2) que se usan comúnmente en estudios de metabarcoding. Sin embargo, estudios recientes sugieren que la construcción de una base de datos de referencia seleccionada y adaptada a los objetivos específicos del estudio puede aumentar la sensibilidad de la clasificación, reducir la tasa de falsos positivos y prevenir redundancia en la clasificación de secuencias por especies (Arranz et al., 2020). Por lo tanto, existen diferentes alternativas para generar bases de datos “personalizadas” a partir de las bases de datos generales. Un ejemplo es la base de datos MARES, la cual es específica para especies eucariotas marinas con identificadores taxonómicos estandarizados y curados por especialistas, que incluye alrededor de 17,884 secuencias del gen mitocondrial que codifica la subunidad I de la enzima citocromo oxidasa (COI) de especies marinas (Arranz et al., 2020). Otra alternativa para la baja asignación taxonómica es el uso de unidades taxonómicas operativas (OTU, por sus siglas en inglés) como hipótesis de trabajo donde las secuencias de referencia no están disponibles, debido a que las secuencias de ADN contienen señales filogenéticas que permiten diferenciar los organismos presentes en una muestra independientemente de su correcta identificación taxonómica (Berry et al., 2015).
Tabla 2
Lista de bases de datos genéticos y genómicos de secuencias de ADN y ARN útiles para la asignación taxonómica y definición de unidades taxonómicas operacionales (OTU).
| Bases de datos | Dirección url | Tipo de datos | País |
| GenBank | https://www.ncbi.nlm.nih.gov/genbank/ | Secuencias de ADN, ARN, SNPs, EST, Genomas completos | Estados Unidos |
| DDBJ center | https://www.ddbj.nig.ac.jp | Secuencias de ADN, ARN, SNPs, EST, Genomas completos | Japón |
| EMBL-EBI | https://www.ebi.ac.uk/ | Secuencias de ADN, ARN, SNPs, EST, Genomas completos | Inglaterra |
| Barcode of Life Data System (BOLD) | https://www.boldsystems.org/index.php | Secuencias de COI (animales), MatK, rbcL (Plantas), ITS (hongos) | Canadá |
| Marine Eukaryote Species (MARES) | https://github.com/wpearman1996/ MARES_database_pipeline | Secuencias de COI de eucariotas marinos | Nueva Zelanda |
| SILVA | https://www.arb-silva.de/ | Base de datos ARN ribosomal de alta calidad | Alemania |
| UNITE | https://unite.ut.ee/ | Base de datos de secuencias ITS de eucariotas, principalmente hongos | Países Nórdicos |
Cuantificación de las lecturas de metabarcoding para el análisis de dieta
El recuento de secuencias se utiliza para registrar datos de presencia de especies dentro de las muestras en función de un número umbral de secuencias (es decir, presencia/ausencia de taxa), o bien para calcular el porcentaje de ADN de cada uno de los ítems-artículos en la dieta, como un aproximado de la biomasa relativa consumida (es decir, la abundancia relativa de secuencias de cada grupo taxonómico respecto al total de secuencias obtenidas en la muestra) (Deagle et al., 2019). La conversión del recuento de secuencias a datos de ocurrencia a menudo se considera un enfoque más conservador, mientras que el uso del recuento de secuencias como indicador de biomasa en muestras es más controvertido. De hecho, las dificultades para obtener una firma de biomasa precisa a partir del recuento de secuencias incluyen sesgos, tanto técnicos como biológicos, que afectan las tasas de recuperación de los marcadores genéticos de diferentes taxones (Deagle et al., 2019). Por lo tanto, en el mejor de los casos, los recuentos de lectura de secuencias solo pueden proporcionar una estimación aproximada de la abundancia proporcional (Deagle et al., 2019).
Con los datos de ocurrencia en lugar de ítem alimenticios de las muestras individuales se pueden usar varias métricas para describir la dieta en las muestras, algunas son el porcentaje de frecuencia de ocurrencia (% FOO), el porcentaje de ocurrencia (POO) y el porcentaje ponderado de ocurrencia (wPOO) (Deagle et al., 2019). Además, algunos estudios también presentan datos de abundancia relativa de lecturas (RRA, por sus siglas en inglés) junto con los datos de presencia (Deagle et al., 2019).
Casos de estudio de dieta en peces
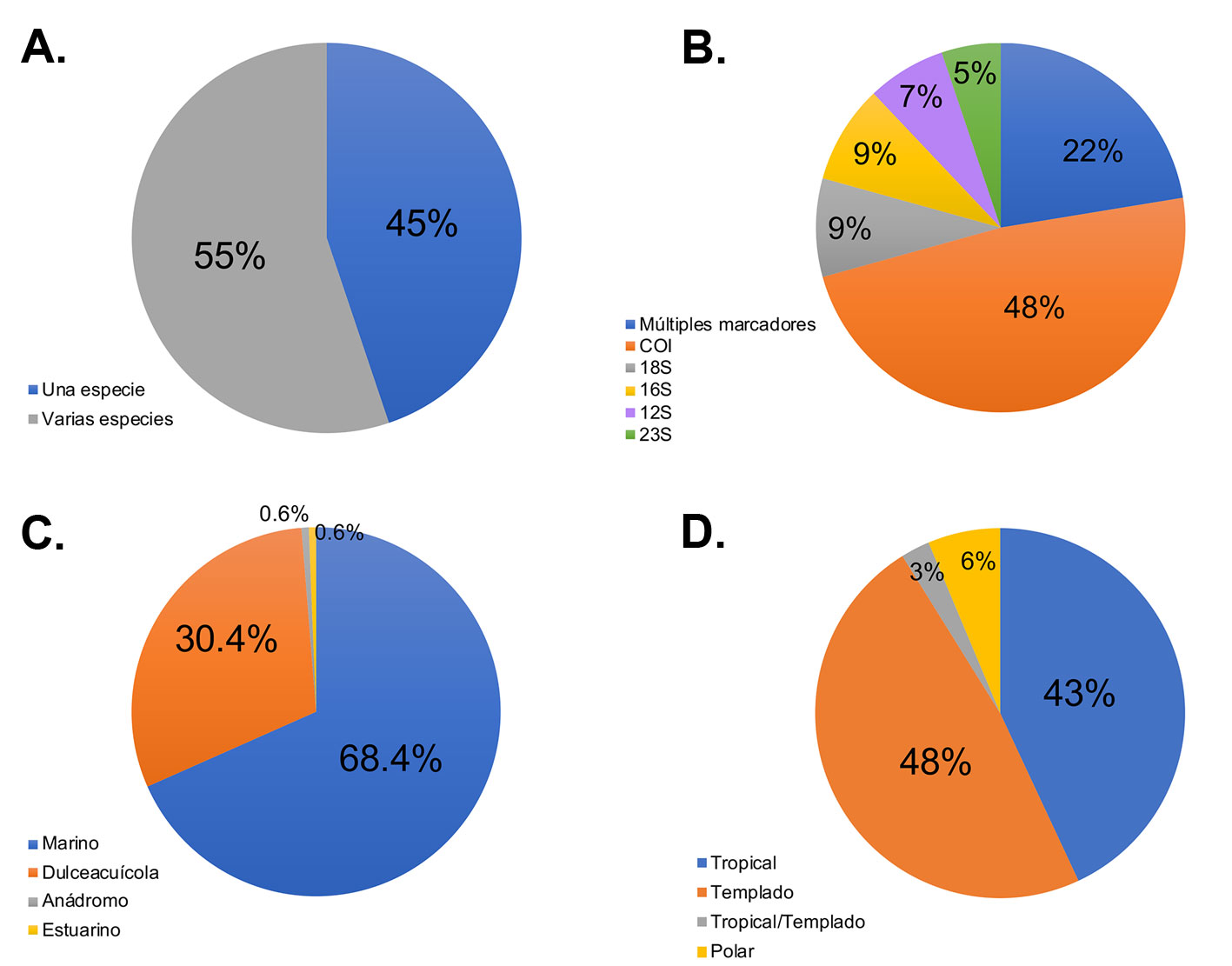
En los últimos años, el uso de herramientas relacionadas con metabarcoding en investigaciones que estudian la dieta de especies de peces se ha multiplicado. En el presente trabajo se realizaron búsquedas y revisión de literatura en Web of Science (http://webofscience.com, última revisión realizada el 11/08/2022), utilizando las palabras clave “metabarcoding”, “fish” y “diet” o “trophic ecology”, las cuales arrojaron como resultado 267 y 131 referencias, respectivamente. Después se revisaron estas referencias para seleccionar específicamente los estudios relacionados con la ecología trófica de peces analizados con herramientas de metabarcoding. De estas búsquedas se obtuvieron un total de 58 artículos enfocados en peces que datan de los últimos 10 años, a partir de los cuales se realizó una síntesis actualizada sobre los avances que se tienen en el tema. De los 58 artículos revisados (material suplementario: tabla S1), 42 fueron publicados en los últimos 5 años, 32 analizaron varias especies de peces de manera simultánea (entre 2 y 22 especies) y 26 fueron monoespecíficos (fig. 2A), para un total de 158 especies analizadas: 1 pez sin mandíbula (familia Petromyzonthidae), 9 peces cartilaginosos (clase Chondrichthyes) y 148 óseos (clase Actinopterygii). Tres estudios utilizaron 3 marcadores genéticos, 10 estudios 2 marcadores, y los restantes (45) 1 solo marcador (fig. 2B). El gen mitocondrial COI fue el marcador más utilizado (28 estudios), seguido de los genes ribosomales 18S (5 estudios), 16S (5 estudios), 12S (4 estudios) y 23S (3 estudios) (fig. 2B). El uso más frecuente del marcador universal COI se puede deber a que: i) es uno de los marcadores moleculares más fácil de amplificar por PCR en el laboratorio y ofrece resultados con menor porcentaje de error en comparación con otros marcadores; ii) presenta una gran precisión taxonómica, identificando las presas del grupo de los invertebrados hasta género y/o especie, que comprenden una alta proporción en la dieta de un gran número de especies; iii) presenta las bases de datos moleculares más completas y depuradas en comparación con otros marcadores (Devloo-Delva et al., 2018; Jakubaviciute et al., 2017). Sin embargo, se ha reportado que los oligonucleótidos cebadores para la amplificación de COI fallan en la detección de grupos taxonómicos como los phyla Platyhelminthes, Nematoda y algunos órdenes de crustáceos, que incluyen varios grupos de la subclase Copepoda, los cuales sí son identificados con los genes ribosomales 18S y 16S (Berry et al., 2015; Devloo-Delva et al., 2018).
Por su parte, los genes ribosomales 12S, 16S y 18S, poseen una capacidad de resolución menor para la identificación taxonómica, llegando a nivel de orden y familia para la mayoría de las presas eucariontes, o de género en casos específicos (principalmente bacterias) (Berry et al., 2015; Hirai et al., 2017). Aun así, el marcador 18S presenta mayor amplitud taxonómica, detectando varios grupos de eucariotas que el marcador COI no tiene el poder de detectar, como protistas, hongos y algunas algas que forman parte de la dieta de un importante número de especies de peces (Hirai et al., 2017), y a su vez, falla en detectar varios grupos de peces y artrópodos (como el orden Amphipoda y el subphylum Hexapoda) que COI sí detecta (Berry et al., 2015). Por su parte, el gen 23S ha permitido detectar con éxito un amplio grupo de especies y géneros de algas, microalgas y cianobacterias que forman parte de la dieta de la mayoría de peces herbívoros (Gajdzik et al., 2021; Nalley et al., 2022; Sherwood y Presting, 2007; Stamoulis et al., 2017). Esta variedad de resultados taxonómicos de los distintos marcadores moleculares ha llevado a que se recomiende la combinación de estos marcadores, ya que esta estrategia arroja información complementaria para la estimación de la riqueza de especies presente en la dieta de los peces (Devloo-Delva et al., 2018), siendo un claro ejemplo de cómo los estudios con más de un marcador metabarcoding describen la dieta con un mayor detalle. Sin embargo, solo 13 de los 58 artículos revisados analizaron múltiples loci, lo cual puede deberse al incremento del costo que implican los análisis multilocus (Devloo-Delva et al., 2018). Los costos de HTS disminuyen paulatinamente, haciendo más factible este tipo de estudios en el futuro.
Es de destacar el uso de los oligonucleótidos cebadores MiFish, los cuales amplifican una región hipervariable de aproximadamente 172 pb del gen ribosomal 12S. Estos oligonucleótidos cebadores que han sido diseñados exclusivamente para la estimación de la diversidad de peces, con los cuales se han podido identificar familias, géneros y especies en muestras ambientales, han sido probados con éxito en ecosistemas marinos, estuarinos y de agua dulce alrededor del mundo (Miya et al., 2015), y han demostrado ser una excelente opción para la estimación de OTU y riqueza de especies de peces en muestras provenientes de contenidos estomacales o heces (Miya et al., 2020), convirtiéndose en una excelente alternativa para el estudio trófico de especies ictiófagas, o cuya dieta está conformada en parte por este ítem-artículo alimenticio, aunque hasta ahora no han sido utilizado en estudios tróficos de peces. En estudios comparativos con otros marcadores metabarcoding como COI, los cebadores MiFish probaron ser más eficientes en la obtención de lecturas de peces, con menor detección en procariotas y otros eucariotas, a pesar de obtener un menor número de lecturas (Collins et al., 2019; Doble et al., 2019). Las desventajas de los cebadores MiFish son la presencia de falsos negativos en algunos grupos como tiburones y rayas, algunas especies de la familia Cotiidae, especies de lampreas y especies taxonómicamente crípticas de caballitos marinos, y aunque se han diseñado cebadores específicos para la detección de estos grupos, su efectividad en estudios de campo aún no ha sido examinada (Miya et al., 2020). Otra desventaja es la falta de variación de los cebadores MiFish para detectar especies congéneres cercanamente relacionadas. Por lo que, para poder discriminar entre las especies cercanas se ha propuesto el uso de cebadores metabarcoding de otros marcadores como ND5, ND2, ATP6 o “Región Control”, junto con los cebadores MiFish en una reacción multiplex (Miya et al., 2020). Es importante resaltar que los cebadores MiFish están siendo usados en los consorcios internacionales como el DNAqua-Net de la Unión Europea y el consorcio creado por varias universidades e institutos que están investigando los peces distribuidos en la corriente de California, para lo cual han creado una base de datos de referencia de secuencias MiFish de especies de peces llamada FishCARD (Miya et al., 2020), paso importante para el desarrollo de la técnica de metabarcoding en estudios tróficos, dada la necesidad de contar con colecciones moleculares de referencia.
Es importante señalar que el análisis de los estudios de peces publicados en los últimos 10 años indica una progresión natural de los tópicos estudiados con la herramienta de metabarcoding. Específicamente, entre 2013-2018, 13 de los 21 artículos publicados se enfocaron en determinar la efectividad de diferentes primers o cebadores (Albaina et al., 2016; Corse et al., 2017; Leray et al., 2013; Yoon et al., 2017), realizar análisis comparativos entre distintos marcadores (Devloo-Delva et al., 2018), probar la eficiencia de los “blocking primers” necesarios para los análisis por metabarcoding (Su et al., 2018), y comparar los resultados obtenidos a partir de métodos de identificación visual, isótopos estables y metabarcoding (Berry et al., 2015; Guillerault et al., 2017; Jakubaviciute et al., 2017; Kodoma et al., 2017; Matley et al., 2018; Oyafuso et al., 2016; Riccioni et al., 2018) (material suplementario: tabla S1). Lo anterior indica que los primeros artículos publicados estuvieron predominantemente enfocados a cuestiones metodológicas necesarias para la aplicación del metabarcoding en el estudio de la ecología trófica de peces. Sin embargo, a partir del año 2019, en 34 de 37 artículos publicados, el método de metabarcoding es usado para responder preguntas ecológicas sobre la especialización trófica y definición del nicho de las especies, la reconstrucción de redes tróficas en comunidades complejas y el efecto del calentamiento global en la dieta, entre otras (material suplementario: tabla S1). Estas investigaciones demuestran que el uso de metabarcoding representa una herramienta valiosa para el estudio de la ecología trófica
de peces.
Respecto a los ecosistemas estudiados, los resultados de la revisión indican que 42 estudios fueron realizados con especies marinas (108 especies), con la inclusión de 1 especie estuarina; 15 con peces dulceacuícolas (48 especies) y 1 con una especie anádroma (fig. 2C). Respecto al tipo de ecosistemas, 22 estudios fueron realizados en peces distribuidos en regiones tropicales (68 especies), 31 estudios en regiones templadas (76 especies), 2 estudios con especies distribuidas en ambas regiones (3 especies dulceacuícolas y 1 marina), y 4 estudios en regiones polares (10 especies) (fig. 2D). Destaca la ausencia de estudios en Latinoamérica (aunque si bien hay 2 estudios, 1 en Puerto Rico y otro en Belice, se realizaron por investigadores extranjeros; Harms-Tuohy et al. [2016] y Brandl et al. [2020]), África y el sur de Asia, las cuales representan regiones de alta diversidad y endemismos.
El uso de la técnica de metabarcoding para el estudio trófico en ecosistemas acuáticos en México
Los peces constituyen el grupo más diverso de vertebrados acuáticos, sin embargo, la mayoría de estudios se han centrado en regiones templadas (Paleártica y Neártica). México se ubica en una zona de transición biogeográfica, de ahí su importancia para el estudio trófico de peces provenientes de ecosistemas templados y tropicales. Además, México es uno de los países más diversos en especies ícticas, con un total de 2,763 especies (505 en ambientes dulceacuícolas y 2,224 en ambientes marinos), 967 géneros, 265 familias y 53 órdenes, que constituyen 9.8% de la totalidad de las especies reconocidas a escala mundial. Además, presenta un alto nivel de endemismos en ambientes dulceacuícolas (57%, correspondiente a 289 especies) y en ambientes marinos como el golfo de California (20%) (Espinosa-Pérez, 2014). Sin embargo, hasta el momento, no se han realizado estudios de ecología trófica mediante metabarcoding con especies de peces distribuidos en México. Este es un reto considerable y atractivo, ya que la identificación de presas y el nivel de resolución taxonómica en los estudios de metabarcoding depende del conocimiento de la biodiversidad y la existencia de secuencias en bases de datos públicas, lo cual implica un muestreo taxonómico preciso de organismos acuáticos de diferentes niveles jerárquicos, con los datos moleculares asociados.
Dado que las cuencas dulceacuícolas son consideradas islas biogeográficas, tener un perfil taxonómico de la diversidad biológica de cada cuenca es un trabajo arduo. Por ejemplo, en México, la Conabio reconoce 27 cuencas continentales distribuidas en 2 ecoregiones (Neártica y Neotropical) (https://www.biodiversidad.gob.mx/region/ecorregiones-dulceacuícolas) y varios estudios que evalúan la distribución taxonómica y genética de organismos dulceacuícolas han encontrado un gran número de endemismos o linajes evolutivos independientes, restringidos a las cuencas o incluso a cuerpos de agua particulares (Ballesteros-Nova et al., 2019; Beltrán-López et al., 2017, 2018; Domínguez-Domínguez et al., 2006; García-Morales et al., 2021; Mercado-Salas et al., 2018; Pedraza-Lara et al., 2012).
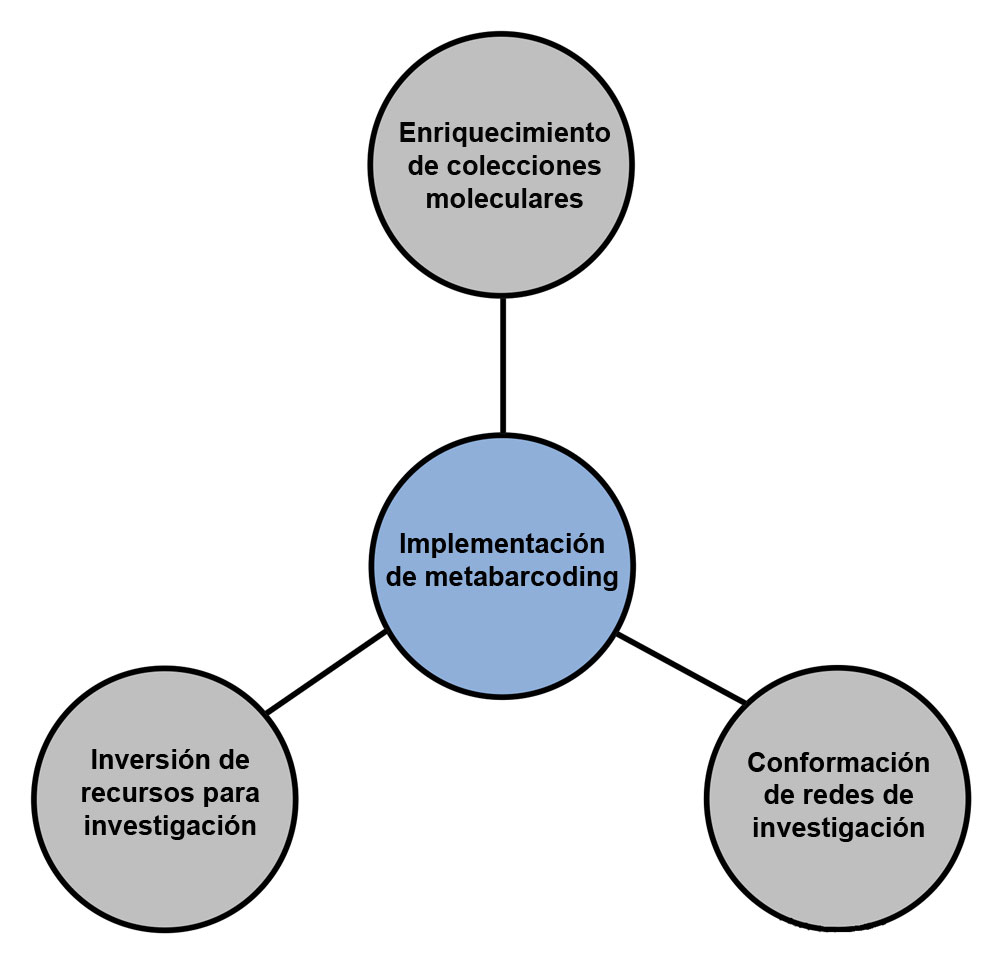
Con el fin de conocer el número de secuencias registradas y curadas de las especies acuáticas que se encuentran distribuidas en el territorio mexicano, en esta revisión se realizó una búsqueda de secuencias en la base de datos BOLD Systems, utilizando el nombre del grupo taxonómico de interés a nivel de clase o phylum, junto con la palabra México (por ejemplo; Actinopterygii+Mexico) (https://www.boldsystems.org/index.php/databases revisada el 2 de noviembre del 2022). De manera general, los resultados de la búsqueda indicaron que los peces óseos (clase Actinopterygii) y los peces cartilaginosos (clase Chondrichthyes), así como la clase Insecta (muchos de los cuales son acuáticos o tiene alguna de sus fases de vida en estos ecosistemas) y las especies acuáticas del phylum Arthropoda (sin incluir la clase Insecta), presentan un avance significativo en el muestreo molecular (tabla 3). Sin embargo, otras especies acuáticas del subphylum Urochordata (acidias y tunicados), familia Petromyzontidae (lampreas), peces de la clase Holocephali (peces Quimeras) y otros taxones de invertebrados menos diversos y estrictamente acuáticos (por ejemplo, Cnidaria, Porifera, Ctenophora, Echinodermata, Rotifera) están pobremente representados, al igual que los grupos de macroalgas (Chlrophyta, Rodophyta y Ochrophyta) y especies de protozoarios acuáticos (superclase Bacillariophyceae y superclase Dinoflagellata) (tabla 3). Por lo anterior, aunque aún se desconoce una gran parte de la biodiversidad presente en los ecosistemas acuáticos de México, esta base de datos constituye un insumo valioso para consolidar investigaciones que implementen técnicas como el metabarcoding, ya que uno de los pasos importantes para consolidar esta técnica, es la construcción de una colección molecular que refleje la diversidad biológica de los ambientes y que se pueda utilizar para la asignación taxonómica de OTU en los análisis metagenómicos (figs. 1, 3).
Lo anterior enfatiza la necesidad de contar con una estrategia científica para la implementación de la técnica de metabarcoding en los estudios que evalúan la diversidad marina y dulceacuícola de México, por ejemplo, la creación de un consorcio o red temática de investigación que fortalezca la colaboración entre diferentes grupos de investigación, universidades e institutos científicos, así como entidades gubernamentales y organizaciones no gubernamentales (fig. 3). Esto permitiría la vinculación de diferentes proyectos de monitoreo (tanto morfológico como molecular) de la diversidad en los sistemas acuáticos en el país y contribuiría al enriquecimiento de las bases de datos (tanto nacionales como las internacionales existentes; tabla 2) de marcadores moleculares (COI, 12S, 16S, 18S y 23S), tal como lo hizo en su momento la red temática MEXBOL (www.mexbol.org). Esta iniciativa requiere del fortalecimiento de la infraestructura física necesaria para la realización de los análisis moleculares en sus diferentes fases (para lo cual existen avances considerables a nivel de laboratorios y equipos en diferentes instituciones del país) y contar con una técnica estandarizada que garantice la comparación de diferentes estudios (fig. 3). También es importante enfocarse en el monitoreo de aquellos marcadores que cuentan con pocos datos (como los genes ribosomales); en los grupos biológicos que históricamente han sido escasamente muestreados; así como en ambientes donde la adaptación y especiación local son frecuentes (como las cuencas hidrográficas continentales o la zona mesofótica marina), o donde la presencia de endemismos es alto (como en el archipiélago de Revillagigedo, el golfo de California, o los lagos del altiplano mexicano).
Tabla 3
Número de secuencias moleculares de diferentes grupos taxonómicos acuáticos que se distribuyen en México y que se encuentran en la base pública del BOLD systems.
| Grupo taxonómico | Registros con secuencias | Registros con nombre de especie | Número de especies* | Secuencias no especificadas |
| Actinopterygii | 10,311 | 7,833 | 922 | 2,478 |
| Elasmobranchii | 351 | 294 | 54 | 57 |
| Hyperoartia | 2 | 2 | 2 | 0 |
| Holocephali | 2 | 0 | 0 | 2 |
| Ascidiacea | 1 | 1 | 1 | 0 |
| Myxini | 0 | 0 | 0 | 0 |
| Insecta | 58,782 | 17,062 | 3,869 | 41,720 |
| Arthropoda sin Insecta | 5,329 | 4,052 | 536 | 1,277 |
| Mollusca | 1,432 | 1,116 | 265 | 316 |
| Annelida | 991 | 431 | 137 | 560 |
| Platyhelminthes | 749 | 688 | 81 | 61 |
| Rotifera | 723 | 635 | 108 | 88 |
| Nematoda | 470 | 193 | 38 | 277 |
| Cnidaria | 388 | 231 | 41 | 157 |
| Echinodermata | 280 | 258 | 83 | 22 |
| Ctenophora | 24 | 21 | 12 | 3 |
| Porifera | 15 | 13 | 13 | 2 |
| Nemertea | 8 | 4 | 2 | 4 |
| Hemichordata | 6 | 2 | 2 | 4 |
| Ciliophora | 6 | 6 | 2 | 0 |
| Phoronida | 4 | 0 | 0 | 4 |
| Nematomorpha | 1 | 1 | 1 | 1 |
| Bryozoa | 0 | 0 | 0 | 0 |
| Placozoa | 0 | 0 | 0 | 0 |
| Priapulida | 0 | 0 | 0 | 0 |
| Rhodophyta | 266 | 175 | 107 | 51 |
| Phaeophyceae | 62 | 62 | 14 | 0 |
| Chlorophyta | 44 | 40 | 26 | 4 |
| Bacillariophyta (diatomeas) | 12 | 12 | 8 | 4 |
| Dinophyceae | 2 | 2 | 2 | 2 |
*Con al menos una secuencia
Finalmente, ante el escenario actual, donde los ecosistemas están viviendo un momento de acelerado deterioro y cambio ecosistémico por las actividades humanas, y cuyas consecuencias sobre las comunidades naturales son aún impredecibles, el uso de técnicas metabarcoding para el estudio de la dieta de los peces se presenta como una área de oportunidad, ya que brinda evidencia directa de los procesos ecosistémicos que rigen el flujo de materia y energía dentro de las redes tróficas acuáticas, así como de las dinámicas ecosistémicas importantes para el mantenimiento de las comunidades biológicas a largo plazo, al permitir la identificación de los ítems alimenticios con mayor resolución taxonómica que los análisis de inspección visual de contenidos estomacales o excretas. Además, las muestras de las dietas son una muestra de la diversidad (también llamadas cápsulas de biodiversidad; de Sousa et al., 2019), que de manera similar a las herramientas moleculares de ADN ambiental, se pueden usar en el monitoreo de la diversidad biológica en los ecosistemas acuáticos, que permite por un lado conocer la ecología trófica de las especies estudiadas, y por otro obtener conocimiento de la biodiversidad de los ecosistemas donde las especies bajo estudio habitan, lo cual en conjunto con otros quehaceres científicos ayudará a dar pautas para la generación de políticas de manejo y conservación de los ecosistemas acuáticos basadas en el conocimiento científico. Esto se vuelve importante en especies usadas en la industria pesquera, ya que conocer la dieta y su papel trófico en la comunidad biológica, permite entender cómo su pesca afecta los ecosistemas acuáticos.
Agradecimientos
El presente estudio fue realizado con apoyo del proyecto PAPIIT-DGAPA-UNAM IN217420 otorgado a NCC. VP agradece al Programa de Becas Posdoctorales DGAPA de la UNAM por la beca otorgada. Los autores agradecen a L. Escalera-Vázquez, a dos árbitros anónimos y al editor de tema por la revisión y comentarios al manuscrito.
Referencias
Albaina, A., Aguirre, M., Abad, D., Santos, M. y Estonba, A. (2016). 18S rRNA V9 metabarcoding for diet characterization: a critical evaluation with two sympatric zooplanktivorous fish species. Ecology and Evolution, 6, 1809–1824. https://doi.org/10.1002/ece3.1986
Alberdi, A., Aizpurua, O., Bohmann, K., Gopalakrishnan, S., Lynggaard, C., Nielsen, M. et al. (2019). Promises and pitfalls of using high-throughput sequencing for diet analysis. Molecular Ecology Resources, 19, 327–348. https://doi.org/10.1111/1755-0998.12960
Anantharaman, K., Brown, C. T., Hug, L. A., Sharon, I., Castelle, C. J., Probst, A. J. et al. (2016). Thousands of microbial genomes shed light on interconnected bio- geochemical processes in an aquifer system. Nature Communications, 7, 13219. https://doi.org/10.1038/ncomms13219
Arranz, V., Pearman, W. S., Aguirre, J. D. y Liggins, L. (2020). MARES, a replicable pipeline and curated reference database for marine eukaryote metabarcoding. Scientific Data, 7, 1–8. https://doi.org/10.1038/s41597-020-0549-9
Bachiller, E., Albo-Puigserver, M., Giménez, J., Pennino, M. G., Marí-Mena, N., Esteban, A. et al. (2020). A trophic latitudinal gradient revealed in anchovy and sardine from Western Mediterranean Sea using a multi-proxy approach. Scientific Reports, 10, 17598. https://doi.org/10.1038/s41598-
020-74602-y
Bachiller, E., Gimenéz, J., Albo-Puigserver, M., Pennino, M. G., Marí-Mena, N., Esteban, A. et al. (2021). Trophic niche overlap between round safinell (Sardinella aurita) and sympatric pelagic fish species in the Western Mediterranean. Ecology and Evolution, 11, 16126–16142. https://doi.org/10.1002/ece3.8293
Ballesteros-Nova, N.E., Pérez-Rodríguez, R., Betrán-López, R.G. y Domínguez-Domínguez, O. (2019). Genetic differentiation in the southern population of the fathead minnow Pimephales promelas rafinesque (Actinopterygii: Cyprinidae). PeerJ, 7, e6224. https://doi.org/10.7717/peerj.6224
Beltrán-López, R. G., Domínguez-Domínguez, O., Guerrero, J. A., Corona-Santiago, D. K., Mejía-Mojica, H. y Doadrio, I. (2017). Phylogeny and taxonomy of the genus Ilyodon Eigenmann, 1907 (Teleostei: Goodeidae), based on mitochondrial and nuclear DNA sequences. Journal of Zoological Systematics and Evolutionary Research, 55, 340–355. https://doi.org/10.1111/jzs.12175
Beltrán-López, R. G., Domínguez-Domínguez, O., Pérez-Rodríguez, R., Kyle, P. y Doadrio, I. (2018). Evolving in the highlands: the case of the Neotropical Lerma live-bearing Poeciliopsis infans (Wolman, 1984) (Cyprinodontiformes: Poeciliidae) in Central Mexico. BMC Evololutionary Biology, 18, 56. https://doi.org/0.1186/s12862-018-1172-7
Berry, O., Bulman, C., Bunce, M., Coghlan, M., Murray, D. C. y Ward, R. D. (2015). Comparison of morphological and DNA metabarcoding analyses of diets in exploited marine fishes. Marine Ecology Progress Series, 540, 167–181. https://doi.org/10.3354/meps11524
Bessey, C., Jarman, S. N., Stat, M., Rohner, C. A., Bunce, M., Koziol, A. et al. (2019). DNA metabarcoding assays reveal a diverse prey assemblage for Mobula rays in the Bohol Sea, Philippines. Ecology and Evolution, 9, 2459–2474. https://doi.org/10.1002/ece3.4858
Bonato, K. O., Silva, P. C., Carvalho, R. F. y Malabarba, L. R. (2022). Trophic interactions of vampire catfishes (Siluriformes: Vandelliinae) revealed by metabarcoding analysis of stomach contents. Freshwater Biology, 67, 542–548. https://doi.org/10.1111/fwb.13861
Bowser, A. K., Diamond, A. W. y Addison, J. A. (2013). From puffins to plankton: A DNA-based analysis of a seabird food chain in the northern Gulf of Maine. Plos One, 8, e83152. https://doi.org/10.1371/journal.pone.0083152
Brandl, S. J., Casey, J. M. y Meyer, C. P. (2020). Dietary and habitat niche partitioning in congeneric cryptobenthic reef fish species. Coral Reefs, 39, 305–317. https://doi.org/10.1007/s00338-020-01892-z
de Bruyn, M., Barbato, M., DiBattista, J. D. y Broadhurst, M. K. Secondary predation constrains DNA-based diet reconstruction in two threatened shark species. Scientific Reports, 11, 18350. https://doi.org/10.1038/s41598-021-96856-w
Bunch, A. J., Carlson, K. B., Hoogakker, F. J., Plough, L. V. y Evans, H. K. (2021). Atlantic sturgeon (Acipenser oxyrinchus oxyrinchus Mitchill, 1815) early life stage consumption evidenced by high-throughput DNA sequencing. Journal of Applied Ichthyology, 37, 12–19. https://doi.org/10.1111/jai.14153
Carlisle, A. B., Andruszkiewicz, A. E., Kim, S. L., Meyer, L., Port, J., Scherrer, S. et al. (2021). Integrating multiple chemical tracers to elucidate the diet and habitat of Cookiecutters sharks. Scientific Reports, 11, 11809. https://doi.org/10.1038/s41598-021-89903-z
Cartes, J. E., Soler-Membrives, A., Stefanescu, C., Lombarte, A. y Carrassón, M. (2016). Contributions of allochthonous inputs of food to the diets of benthopelagic fish over the northwest Mediterranean slope (to 2300 m). Deep Sea Research Part I: Oceanographic Research Papers, 109, 123–136. https://doi.org/10.1016/j.dsr.2015.11.001
Casey, J. M., Meyer, C. P., Morat, F., Brandl, S. J., Planes, S. y Parravicini, V. (2019). Reconstructing hyper diverse food webs: gut content metabarcoding as a tool to disentangle trophic interactions on coral reefs. Methods in Ecology and Evolution, 10, 1157–1170. https://doi.org/10.1111/2041-210X.13206
Castro-Sanguino, C. y Sánchez, J. A. (2012). Dispersal of Symbiodinium by the stoplight parrotfish Sparisoma viride. Biology Letters, 8, 282–286. https://doi.org/10.1098/rsbl.2011.0836
CBOL, P. W. G. (2009). A DNA barcode for land plants. Proceedings of the National Academy of Sciences, 106, 12794–12797. https://doi.org/10.1073/pnas.0905845106
Clarke, L. J., Trebilco, R., Walters, A., Polanowski, A. M. y Deagle, B. E. (2020). DNA-based diet analysis of mesopelagic fish from the southern Kerguelen Axis. Deep Sea Research Part II, 174, 104494. https://doi.org/10.1016/j.dsr2.2018.09.001
Collins, R. A., Bakker, J., Wangensteen, O. S., Soto, A. Z., Corrigan, L., Sims, D. W. et al. (2019). Non-specific amplification compromises environmental DNA metabarcoding with COI. Methods in Ecology and Evolution, 10, 1985–2001. https://doi.org/10.1111/2041-210X.13276
Corse, E., Meglècz, E., Archambaud, G., Ardisson, M., Martin, J. F., Tugard, C. et al. (2017). A from-benchtop-to-desktop workflow for validating HTS data and for taxonomic identification in diet metabarcoding studies. Molecular Ecology Resources, 17, e146–e159. https://doi.org/10.1111/1755-0998.12703
Corse, E., Tougard, C., Archambaud-Suard, G., Agnèse, J. F., Messu-Mandeng, F. D., Bilong-Bilong, C. F. et al. (2019). One-locus-several-primers: a strategy to improve the taxonomic and haplotypic coverage in diet metabarcoding studies. Ecology and Evolution, 9, 4603–4620. https://doi.org/10.1002/ece3.5063
Creer, S., Deiner, K., Frey, S., Porazinska, D., Taberlet, P., Thomas, W. K. et al. (2016). The ecologist´s field guide to sequence-based identification of biodiversity. Methods in Ecology and Evolution, 7, 1008–1018. https://doi.org/10.1111/2041-210X.12574
Cuff, J. P., Windsor, F. M., Tercel, M. P. T. G., Kitson, J. J. N. y Evans, D. M. (2022). Overcoming the pitfalls of merging dietary metabarcoding into ecological networks. Methods in Ecology and Evolution, 13, 545–559. https://doi.org/10.1111/2041-210X.13796
da Silveira, E. L., Semmar, N., Cartes, J. E., Tuset, V. M., Lombarte, A., Ballester, E. L. C. et al. (2020). Methods for trophic ecology assessment in fishes: a critical review of stomach analyses. Reviews in Fisheries Science y Aquaculture, 28, 71–106. https://doi.org/10.1080/23308249.2019.1678013
Dahl, K. A., Patterson, W. F., Robertson, A. y Ortmann, A. C. (2017). DNA barcoding significantly improves resolution of invasive lionfish diet in the Northern Gulf of Mexico. Biological Invasions, 19, 1917–1933. https://doi.org/10.1007/s10530-017-1407-3
Deagle, B. E., Eveson, J. P. y Jarman, S. N. (2006). Quantification of damage in DNA recovered from highly degraded samples – a case study on DNA in feces. Frontiers in Zoology, 3, 11. https://doi.org/10.1186/1742-9994-3-11
Deagle, B. E., Jarman, S. N., Coissac, E., Pompanon, F. y Taberlet, P. (2014). DNA metarbocoding and the cytochrome C oxidase subunit I marker: not a perfect match. Biology Letters, 10, 20140562. https://doi.org/10.1098/rsbl.2014.0562
Deagle, B. E., Thomas, A. C., McInnes, J. C., Clarke, L. J., Vesterinen, E. J., Clare, E. L. et al. (2019). Counting with DNA in metabarcoding studies: How should we convert sequence reads to dietary data? Molecular Ecology, 28, 391–406. https://doi.org/10.1111/mec.14734
de Sousa, L. L., Xavier, R., Costa, V., Humpries, E., Trueman, C., Rosa, R. et al. (2016). DNA barcoding identifies a cosmopolitan diet in the ocean sunfish. Scientific Reports, 6, 28762. https://doi.org/10.1038/srep28762
de Sousa, L. L., Silva, S. M. y Xavier, R. (2019). DNA metabarcoding in diet studies: unveiling ecological aspects in aquatic and terrestrial ecosystems. Environmental DNA, 1, 199–214. https://doi.org/10.1002/edn3.27
Devloo-Delva, F., Huerlimann, R., Chau, G., Matley, J. K., Heupel, M. R., Simpfendorfer, C. A. et al. (2018). How does marker choice affect your diet analysis: comparing genetic markers and digestion levels for diet metabarcoding of tropical-reef piscivores? Marine and Freshwater Research, 70, 8–18. https://doi.org/10.1071/MF17209
Dickie, I. A., Boyer, S., Buckley, H. L., Duncan, R. P., Gardner, P. P., Hogg, I. D. et al. (2018). Towards robust and repeatable sampling methods in eDNA-based studies. Molecular Ecology Resources, 18, 940–952. https://doi.org/10.1111/1755-0998.12907
Doble, C. J., Hipperson, H., Salzburger, W., Horsburgh, G. J., Mwita, C., Murrell, D. J. et al. (2019). Testing the performance of environmental DNA metabarcoding for surveying highly diverse tropical fish communities: a case study from Lake Tanganyika. Environmental DNA, 2, 24–41. https://doi.org/10.1002/edn3.43
Domínguez-Domínguez, O., Doadrio, I. y Pérez-Ponce de León, G. (2006). Historical biogeography of some river basins in central Mexico evidenced by their goodeine freshwater fishes: a preliminary hypothesis using secondary Brooks parsimony analysis. Journal of Biogeography, 33, 1437–1447. https://doi.org/10.1111/j.1365-2699.2006.01526.x
Ducotterd, C., Crovadore, J., Lefort, F., Rubin, J. F. y Ursenbacher, S. (2021). A powerful long metabarcoding method for the determination of complex diet from fecal analysis of the European pond turtle (Emys orbicularis, L. 1758). Molecular Ecology Resources, 21, 433–447. https://doi.org/10.1111/1755-0998.13277
Elbrecht, V., Taberlet, P., Dejean, T., Valentini, A., Usseglio-Polatera, P., Beisel, J. N. et al. (2016). Testing the potential of a ribosomal 16S marker for DNA metabarcoding of insects. PeerJ, 4, e1966. https://doi.org/10.7287/peerj.preprints.1855v1
Elbrecht, V. y Leese, F. (2017). Validation and development of COI metabarcoding primers for freshwater macroinvertebrate bioassessment. Frontiers in Environmental Science, 5, 11. https://doi.org/10.3389/fenvs.2017.00011
Elbrecht, V., Braukmann, T. W. A., Ivanova, N. V., Prosser, S. W. J., Hajbabaei, M., Wright, M. et al. (2019). Validation of COI metabarcoding primers for terrestrial arthropods. PeerJ, 7, e7745. https://doi.org/10.7717/peerj.7745
Espinosa-Pérez, H. (2014). Biodiversidad de peces en México. Revista Mexicana de Biodiversidad, 85 (Supl.), S450–S459. https://doi.org/10.7550/rmb.32264
Evans, H. K., Bunch, A. J., Schmitt, J. D., Hoogakker, F. J. y Carlson, K. B. (2021). High-throughput sequencing outperforms traditional morphological methods in Blue Catfish diet analysis and reveals novel insights into diet ecology. Ecology and Evolution, 11, 5584–5597. https://doi.org/10.1002/ece3.7460
Finlay, J. C., Doucett, R. R. y McNeely, C. (2010). Tracing energy flow in stream food webs using stable isotopes of hydrogen. Freshwater Biology, 55, 941–951. https://doi.org/10.1111/j.1365-2427.2009.02327.x
Folmer, O., Black, M., Hoeh, W., Lutz, R. y Vrijenhoek, R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3, 294–299.
Freeland, J. R. (2017). The importance of molecular markers and primer design when characterizing biodiversity from environmental DNA. Genome/National Research Council Canada = Genome/Conseil National De Recherches Canada, 60, 358–374. https://doi.org/10.1139/gen-2016-0100
French, B. J., Lim, Y. W., Zglicznski, B. J., Edwards, R. A., Rohwer, F. y Sandin, S. A. (2020). Decoding diversity in a coral reef fish species complex with restricted range using metagenomic sequencing of gut contents. Ecology and Evolution, 10, 3413–3423. https://doi.org/10.1002/ece3.6138
Gajdzik, L., DeCarlo, T. M., Koziol, A., Mousavi-Derazmahalleh, M., Coghlan, M., Power, M. W. et al. (2021). Climate-assisted persistence of topical fish vagrants in temperate marine ecosystems. Communications Biology, 4, 1231. https://doi.org/10.1038/s42003-021-02733-7
Galan, M., Pons, J. B., Tournayre, O., Pierre, E., Leuchtmann, M., Pontier, D. et al. (2018). Metabarcoding for the parallel identification of several hundred predators and their preys: application to bat species diet analysis. Molecular Ecology Resources, 18, 474–489. https://doi.org/10.1101/155721
Gallet, A., Koubbi, P., Léger, N., Scheifler, M., Ruiz-Rofriguez, M., Suzuki, M. T. et al. (2019). Low-diversity bacterial microbiota in Southern Ocean representatives of lanternfish general Electrona, Protomyctophum and Gymnoscopelus (family Myctophidae). Plos One, 14, e0226159. https://doi.org/10.1371/journal.pone.0226159
García-Morales, A. E., Domínguez-Domínguez, O. y Elías-Gutiérrez, M. (2021). Uncovering hidden diversity; three new species of the Keratella genus (Rotifera, Monogononta, Brachionidae) of high-altitude water systems from central Mexico. Diversity, 13, 676. https://doi.org/10.3390/d13120676
Gillet, F., Tiouchichine M. L., Galan, M., Blanc, F., Némoz, M., Aulagnier, S. et al. (2015). A new method to identify the endangered Pyrenean desman (Galemys pyrenaicus) and study its diet, using next generation sequencing from feces. Mammalian Biology, 80, 505–509. https://doi.org/0.1016/j.mambio.2015.08.002
Guillerault, N., Bouletreau, S., Iribar, A., Valentini, A. y Santoul, F. (2017). Application of DNA metabarcoding of feces to identify European catfish Silurus glanis diet. Journal of Fish Biology, 90, 2214–2219. https://doi.org/10.1111/jfb.13294
Günther, B., Fromentin, J. M., Metral, L. y Arnaud-Haond, S. (2021). Metabarcoding confirms the opportunistic foraging behavior of Atlantic bluefin tuna and reveals the importance of gelatinous prey. PeerJ, 9, e111757. https://doi.org/10.7717/peerj.11757
Hebert, P. D. N., Ratnasingham, S. y deWaard, J. R. (2003). Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proceedings of the Royal Society of London B: Biological Sciences, 270 (Supl. 1), S96–S99. https://doi.org/10.1098/rsbl.2003.0025
Hajibabaei, M., Singer, G. A. C., Hebert, P. D. N. y Hickey, D. A. (2007). DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends in Genetics, 23, 167–172. https://doi.org/10.1016/j.tig.2007.02.001
Heindler, F. M., Maes, G. E., Delerue-Ricard, S., Bavière, A. Vanden, Hostens, K. y Volckaert, F. A. M. (2019). Diet composition and gut microbiome of 0-group European plaice Pleuronectes platessa L. – Strong homogeneity and subtle spatial and temporal differences. Journal of Sea Research, 144, 67–77. https://doi.org/10.1016/j.seares.2018.11.004
Hirai, J., Hidaka, K., Nagai, S. y Ichikawa, T. (2017). Molecular-based diet analysis of the early postlarvae of Japanese sardine Sardinops melanostictus and Pacific round herring Etrumeus teres. Marine Ecology Progress Series, 564, 99–113. https://doi.org/10.3354/meps12008
Hoenig, B. D, Trevelline, B. K., Nuttle, T. y Porter, B. A. (2021). Dietary DNA metabarcoding reveals seasonal trophic changes among three syntopic freshwater trout species. Freshwater Biology, 66, 509–523. https://doi.org/10.1111/fwb.13656
Hunter, E., Taylor, N., Fox, C. J., Maillard, M., y Taylor, M. I. (2012). Effectiveness of TaqMan probes for detection of fish eggs and larvae in the stomach contents of a teleost predator. Journal of Fish Biology, 81, 320–328. https://doi.org/10.1111/j.1095-8649.2012.03298.x
Ivanova, N., Fazekas, A. J. y Hebert, P. D. N. (2008). Semi-automated, membrane-based protocol for DNA isolation from plants. Plant Molecular Biology Reports, 26, 186–198. https://doi.org/10.1007/s11105-008-0029-4
Jakubaviciute, E., Bergström, U., Eklöf, J. S. Haenel, Q. y Bourlat, S. J. (2017). DNA metabarcoding reveals diverse diet of the three-spinned stickleback in a coastal ecosystem. Plos One, 12, e0186929. https://doi.org/10.1371/journal.pone.0186929
Jedlicka, J. A., Sharma, A. M. y Almeida, R. P. P. (2013). Molecular tools reveal diets of insectivorous birds from predator fecal matter. Conservation Genetics Resources, 5, 879–885. https://doi.org/10.1007/s12686-013-9900-1
Johnson, N. S., Lewandoski, S. A y Merkes, C. (2021). Assessment of sea lamprey (Petromyzon marinus) diet using DNA metabarcoding of feces. Ecological Indicators, 125, 107605. https://doi.org/10.1016/j.ecolind.2021.107605
Juen, A. y Traugott, M. (2006). Amplification facilitators and multiplex PCR: tools to overcome PCR-inhibition in DNA-gut-content analysis of soil-living invertebrates. Soil Biology and Biochemistry, 38, 1872–1879. https://doi.org/10.1016/j.soilbio.2005.11.034
Jungbluth, M. J., Burns, J., Grimaldo, L., Slaughter, A., Katla, A. y Kimmerer, W. (2021). Feeding habits and novel prey of larval fishes in the northern San Francisco Estuary. Environmental DNA, 3, 1059–1080. https://doi.org/10.1002/edn3.226
Jusino, M. A., Banik, M. T., Palmer, J. M., Wray, A. K., Xiao, L., Pelton, E. et al. (2018). An improved method for utilizing high-through- put amplicon sequencing to determine the diets of insectivorous animals. Molecular Ecology Resources, 19, 176–190. https://doi.org/10.1111/1755-0998.12951
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M. et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research, 41, e1. https://doi.org/10.1093/nar/gks808
Kim, M. J., Kim, H. W., Lee, S. R., Kim, N. Y., Lee, Y. J., Joo, H. T. et al. (2022). Feeding strategy of the wild Korean earhorse (Hippocampus haema). Journal of Marine Science and Engineering, 10, 357. https://doi.org/10.3390/jmse10030357
Kocher, T. D., Thomas, W. K., Meyer, A., Edwards, S. V., Pääbo, S., Villablanca, F. X. et al. (1989). Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proceedings if the National Academy of Sciences of the United States of America, 86, 6196–6200. https://doi.org/10.1073/pnas.86.16.6196
Kodoma, T., Hirai, J., Tamura, S., Takhashi, T., Tanaka, Y., Ishihara, T. et al. (2017). Diet composition and feeding habitats of larval Pacific bluefin tuna Thunnus orientalis in the Sea of Japan: integrated morphological and metagenetic analysis. Marine Ecology Progress Series, 583, 211–226. https://doi.org/10.3354/meps12341
Kume, G., Kobari, T., Hirai, J., Kuroda, H., Takeda, T., Ichinomiya, M. et al. (2021). Diet niche segregation of co-occurring larval stages of mesopelagic and commercially important fishes in the Osumi Strait assessed through morphological, ADN metabarcoding, and stable isotope analyses. Marine Biology, 168, 6. https://doi.org/10.1007/s00227-020-03810-x
Lazic, T., Corriero, G., Balech, B., Cardone, F., Deflorio, M., Fosso, B. et al. (2021). Evaluating DNA metabarcoding to analyze diet composition of wild long-snouted seahorse Hippocampus guttulatus. En International Workshop on Metrology for the Sea; learning to measure sea health parameters (Metro Sea); IEEE: Reggio Calabria, Italy, 2021; pp. 257–261. https://doi.org/10.1109/MetroSea52177.2021.9611570
Lee, C. I., Wang, F. Y., Liu, M. Y., Chou, T. K. y Liao, T. Y. (2021). DNA metabarcoding for dietary analysis of Holland´s carp (Spinibarbus hollandi) to evaluate the threat to native fishes in Taiwan. Journal of Fish Biology, 99, 1668–1676. https://doi.org/10.1111/jfb.14875
Leray, M., Yang, J. Y., Meyer, C. P., Mills, S. C., Agudelo, N., Ranwez, V. et al. (2013). A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: applications for characterizing coral reef fish gut contents. Frontiers in Zoology, 10, 34. https://doi.org/10.1186/1742-9994-10-34
Leray, M., Meyer, C. P. y Mills, S. C. (2015). Metabarcoding dietary analysis of coral dwelling predatory fish demonstrates the minor contribution of coral mutualists to their highly partitioned, generalist diet. PeerJ, 3, e1047. https://doi.org/10.7717/peerj.1047
Link, S. J. (2002). Ecological consideration in fisheries management: When does it matter? Fisheries, 27, 10–17. https://doi.org/10.1577/1548-8446(2002)027<0010:ECIFM>2.0.CO;2
Lui, C., Qi, R. J., Jiang, J. Z., Zhang, M. Q. y Wang, J. Y. (2019). Development of a blocking primer to inhibit the PCR amplification of the 18S rDNA sequences of Litipenaeus vannamei and its efficacy in Crassostrea honkongensis. Fronteirs in Microbiology, 10, 830. https://doi.org/10.3389/fmicb.2019.00830
Majaneva, M., Diserud, O. H., Eagle, S. H. C., Hajibabaei, M. y Ekrem, T. (2018). Choice of DNA extraction method affects DNA metabarcoding of unsorted invertebrate bulk samples. Metabarcoding and Metagenomics, 2, e26664. https://doi.org/10.3897/mbmg.2.26664
Malviya, S., Scalco, E., Audic, S., Vincent, F., Veluchamy, A., Poulain, J. et al. (2016). Insights into global diatom distribution and diversity in the world’s ocean. Proceedings of the National Academy of Sciences of the United States of America, 113, E1516-E1525. https://doi.org/10.1073/pnas.1509523113
Matley, J. K. Maes, G. E., Devloo-Delva, F., Huerlimann, R., Chua, G., Tobin, A. J. et al. (2018). Integrating complementary methods to improve diet analysis in fishery-targeted species. Ecology and Evolution, 8, 9503–9515. https://doi.org/10.1002/ece3.4456
Mercado-Salas, N. F., Khodami, S., Kihara, T. C., Elías-Gutiérez, M. y Martínez Arbizu, P. (2018). Genetic structure and distributional patterns of the genus Mastigodiaptomus (Copepoda) in Mexico, with the description of a new species from the Yucatán Peninsula. Arthropod Systematics and Phylogeny, 761, 487–507.
Meyer, M. y Kircher, M. (2010). Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protocols, 6, pdb.prot5448. https://doi.org/10.1101/pdb.prot5448
Minamoto, T., Yamanaka, H., Takahara, T., Honjo, M. N. y Kawabata, Z. (2012). Surveillance of fish species composition using environmental DNA. Limnology, 13, 193–197. https://doi.org/10.1007/s10201-011-0362-4
Miya, M., Sato, Y., Fukunaga, T., Sado, T., Poulsen, J. Y., Sato, K. et al. (2015). MiFish, a set of universal PCR primers for metabarcoding environmental from fishes: detection of more than 230 subtropical marine species. Royal Society Open Science, 2, 150088. https://doi.org/0.1098/rsos.150088
Miya, M., Gotoh, R. O. y Sado, T. (2020). MiFish metabarcoding: a high-throughput approach for simultaneous detection of multiple fish species from environmental and other samples. Fisheries Science, 86, 1–32. https://doi.org/10.1007/s12562-020-01461-x
Mychek-Londer, J. G., Chaganti, S. R. y Heath, D. D. (2020). Metabarcoding of native and invasive species in stomach contents of Great Lakes fishes. Plos One, 15, e0236077. https://doi.org/10.1371/journal.pone.0236077
Nalley, E. M., Sonahue, M. J. y Toonen, R. J. (2022) Metabarcoding as a tool to examine cryptic algae in the diets of two common grazing surgeonfishes, Acanthurus triostegus and A. nigrofuscus. Environmental DNA, 4, 135–146. https://doi.org/10.1002/edn3.206
Nalley, E. M., Donahue, M. J., Heeman, A. y Toonen, R. J. (2022). Quantifying the diet diversity of herbivorous coral reef fishes using systematic review and DNA metabarcoding. Environmental DNA, 4, 191–205. https://doi.org/10.1002/edn3.247
Novillo, M., Desvignes, T., Moreira, E. y Barrera-Oro, E. (2022). Egg predation in Antarctic fish: the ingestion by Notothenia coriiceps of an entire Trematomus brenacchii spawn identified by molecular techniques. Estuarine, Coastal and Shelf Science, 266, 107742. https://doi.org/10.1016/j.ecss.2022.107742
Oehm, J., Juen, A., Nagiller, K., Neuhauser, S. y Traugott, M. (2011). Molecular scatology: How to improve prey DNA detection success in avian faeces? Molecular Ecology Resources, 11, 620–628. https://doi.org/10.1111/j.1755-0998.2011.03001.x
Oyafuso, Z. S., Toonen, R. J. y Franklin, C. E. (2016). Temporal and spatial trends in prey composition of wahoo Acanthocybium solandri: a diet analysis from the central North Pacific Ocean usin visual and DNA bar-coding techniques. Journal of Fish Biology, 88, 1501–1523. https://doi.org/10.1111/jfb.12928
Palumbi, S. (1996) Nucleic acids II: polymerase chain reaction. En D. Hillis, C. Moritz y B. Mable (Eds.), Molecular systematics, 2nd Ed. (pp. 205–247). Sunderland, Massachusetts: Sinauer Associates, Inc.
Pandey, R. N., Adams, R. P. y Flournoy, L. E. (1996). Inhibition of ran- dom amplified polymorphic DNAs (RAPDs) by plant polysaccharides. Plant Molecular Biology Reporter/ISPMB, 14, 17–22. https://doi.org/10.1007/BF02671898
Panteli, N., Mastoraki, M., Nikouli, E., Lazarina, M., Antonopoulou, E. y Kormas, K. A. (2020). Imprinting statistically sound conclusions for gut microbiota in comparative animal studies: a case study with diet and teleost fishes. Comparative Biochemistry and Physiology-Part D, 36, 100738. https://doi.org/10.1016/j.cbd.2020.100738
Pedraza-Lara, C., Doadrio, I., Breinholt, J. W. y Crandall, K. A. (2012). Phylogeny and evolutionary patterns in the dwarf crayfish subfamily (Decapoda: Cambarellinae). Plos One, 7, e48233. https://doi.org/10.1371/journal.pone.0048233
Piñol, J., San Andrés, V., Clare, E. L., Mir, G. y Symondson, W. O. C. (2014). A pragmatic approach to the analysis of diets of generalist predators: The use of next-generation sequencing with no blocking probes. Molecular Ecology Resources, 1, 18–26. https://doi.org/10.1111/1755-0998.12156
Piñol, J., Senar, M. A. y Symondson, W. O. C. (2019). The choice of universal primers and the characteristics of the species mixture determine when DNA metabarcoding can be quantitative. Molecular Ecology, 28, 407-419. https://doi.org/10.1111/mec.14776
Rees, G. N., Shackleton, M. E., Watson, G. O., Dwyer, G. K. y Stoffels, R. J. (2019). Metabarcoding demonstrates dietary niche partitioning in two coexisting blackfish species. Marine and Freshwater Research, 71, 512–517. https://doi.org/10.1071/MF18491
Riaz, T., Shehzad, W., Viari, A., Pompanon, F., Taberlet, P. y Coissac, E. (2011). ecoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Research, 39, e145. https://doi.org/10.1093/nar/gkr732
Riccioni, G., Stagioni, M., Piccinetti, C. y Libralato, S. (2018). A metabarcording approach for the feeding habits of European hake in the Adriatic Sea. Ecology and Evolution, 8, 10435–10447. https://doi.org/10.1002/ece3.4500
Rimoldi, S., Torrecillas, S., Montero, D., Gini, E., Makol, A., Valdenegro, V. et al. (2020). Assessment of dietary supplementation with galactomannan oligosaccharides and phytogenics on gut microbiota of European sea bass (Dicentrarchus labrax) fed low fishmeal and fish oil based diet. Plos One, 15, e0231494. https://doi.org/10.1371/journal.pone.0231494
Sambrook, J., Fritsch, E. F. y Maniatis, T. J. (1989). Molecular cloning: a laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press.
Sanchez-Peregrin, L. (2017). Study of fish assemblages in Ecklonia radiata dominated rock reefs in subtropical eastern Australia and dietary analyses of three species of generalist predators of the family Labridae (Tesis de maestría). School of Environment, Science and Engineering, Southern Cross University. Lismore, Australia.
Sang, T., Crawford, D. J. y Stuessy, T. F. (1997). Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). American Journal of Botany, 84, 1120–1136.
Schrader, C., Schielke, A., Ellerbroek, L. y Johne, R. (2012). PCR inhibitors – occurrence, properties and removal. Journal of Applied Microbiology, 113, 1014-1026. https://doi.org/10.1111/j.1365-2672.2012.05384.x
Sherwood, A. y Presting, G. (2007). Universal primers amplify a 23S rDNA plastid marker in eukaryotic algae and cyanobacteria. Journal of Phycology, 43, 605–608. https://doi.org/10.1111/j.1529-8817.2007.00341.x
Shink, K. G., Sutton, T. M., Murphy, J. M. y López, J. A. (2019). Utilizing DNA metabarcoding to characterize the diet of marine-phase Artic lamprey (Lethenteron camtschaticum) in the eastern Bering Sea. Canadian Journal of Fisheries and Aquatic Sciences, 76, 1993–2002. https://doi.org/10.1139/cjfas-2018-0299
Siegenthaler, A., Wangensteen, O.S., Benvenuto, C., Campos, J. y Mariani, S. (2019). DNA metabarcoding unveils multiscale trophic variation in a widespread coastal opportunist. Molecular Ecology, 28, 232–249. https://doi.org/10.1111/mec.14886
Stamoulis, K. A., Friedlander, A. M., Meywe, C. G., Fernandez-Silva, I. y Toonen, R. J. (2017). Coral reef grazer-benthos dynamics complicated by invasive algae in a small marine reserve. Scientific Reports, 7, 43819. https://doi.org/10.1038/srep43819
Stevens, J. D., Bonfil, R., Dulvy, N. K. y Walker, P. A. (2000). The effects of fishing on sharks, rays, and chimeras (chondrichthyans), and the implications for marine ecosystems. ICES Journal of Marine Science, 57, 476–494. https://doi.org/10.1006/jmsc.2000.0724
Steward, S. D., Kelly, D., Biessy, L., Laroche, O. y Wood, S. A. (2021). Individual diet specialization drives population niche responses to environmental changes in a predator fish population. Food Webs, 27, e00193. https://doi.org/10.1016/j.fooweb.2021.e00193
Su, M., Liu, H., Liang, X., Gui, L. y Zhang, J. (2018). Dietary analysis of marine fish species: enhancing the detection of prey-specific DNA sequences via high-throughput sequencing using blocking primers. Estuaries and Coasts, 41, 560–571. https://doi.org/10.1007/s12237-017-0279-1
Taberlet, P., Coissac, E., Hajibabaei, M. y Rieseberg, L. H. (2012). Environmental DNA. Molecular Ecology, 21, 1789–1793. https://doi.org/10.1111/j.1365-294X.2012.05542.x
Taberlet, P., Bonin, A., Zinger, L. y Coissac, E. (2018). Environmental DNA: for biodiversity research and monitoring. Londres: Oxford University Press. https://doi.org/10.1093/oso/9780198767220.001.0001
Takahashi, M., DiBattista, J. D., Jarman, S., Newman, S. J., Wakefield C. B., Harvey, E. S. et al. (2020). Partitioning of diet between species and life history stages of sympatric and cryptic snappers (Lutjunidae) based on DNA metabarcoding. Scientific Reports, 10, 4319. https://doi.org/10.1038/s41598-020-60779-9
Tate, J. A. y Simpson, B. (2003). Paraphyly of Tarasa (Malvaceae) and diverse origins of the polyploid species. Systematic Botany, 28, 723–737. https://doi.org/10.1043/02-64.1
Tournayre, O., Leuchtmann, M., Filippi-Codaccioni, O., Trillat, M., Piry, S., Pontier, D. et al. (2020). In silico and empirical evaluation of twelve metabarcoding primer set for insectivorous diet analyses. Ecology and Evolution, 10, 6310–6332. https://doi.org/10.1002/ece3.6362
Traugott, M., Thalinger, B., Wallinger, C. y Sint, D. (2021). Fish as predators and prey: DNA-based assessment of their role in food webs. Journal of Fish Biology, 98, 367–382. https://doi.org/10.1111/jfb.14400
Valentini, A., Taberlet, P., Miaud, C., Civade, R., Herder, J., Thomsen, P. F. et al. (2016). Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Molecular Ecology, 25, 929–942. https://doi.org/10.1111/mec.13428
Vamos, E., Elbrecht, V. y Leese, F. (2017). Short COI markers for freshwater macroinvertebrate metabarcoding. Metabarcoding and Metagenomics, 1, e14625. https://doi.org/10.3897/mbmg.1.14625
Verma, S. K. y Singh, L. (2003). Novel universal primers establish identity of an enormous number of animal species for forensic application. Molecular Ecology Notes, 3, 28–31. https://doi.org/10.1046/j.1471-8286.2003.00340.x
Vesterinen, E. J., Ruokoleinen, L., Wahlberg, N., Peña, C., Roslin, T, Laine, V. N. et al. (2016). What you need is what you eat? Prey selection by the bat Myotis daubentonii. Molecular Ecology, 25, 1581–1594. https://doi.org/10.1111/mec.13564
Vestheim, H. y Jarman, S. N. (2008) Blocking primers to enhance PCR amplification of rare sequences in mixed samples- a case study on prey DNA in Antarctic krill stomach. Frontiers in Zoology, 5, 12. https://doi.org/10.1186/1742-9994-5-12
Villsen, K., Corse, E., Meglécz, E., Archambaud-Suard, G., Vignes, H., Ereskovsky, A. V. et al. (2021). DNA metabarcoding reveals adaptive seasonal variation of individual trophic traits in a critically endangered fish. Molecular Ecology, 31, 5889–5908. https://doi.org/10.1101/2021.01.25.428043
van der Walt, A. J., van Goethem, M. W., Ramond, J. B., Makhalanyane, T. P., Reva, O. y Cowan, D. A. (2017). Assembling metagenomes, one community at a time. BioMed Central Genomics, 18, 521. https://doi.org/10.1186/s12864-017-3918-9
van Zinnicq-Bergmann, M. P. M., Postaire, B. D., Gastrich, K., Heithaus, M. R., Hoopes, I. A., Lyons, K. et al. (2021). Elucidating shark diet with DNA metabarcoding from cloacal swabs. Molecular Ecology Resources, 21, 1056–1067. https://doi.org/10.1111/1755-0998.13315
Wangensteen, O. S., Palacín, C., Guardiola, M. y Turon, X. (2018). DNA metabarcoding of littoral hard-bottom communities: high diversity and database gaps revealed by two molecular markers. PeerJ, 6, e4705. https://doi.org/10.7717/peerj.4705
Waraniak, J. M., March, T. L. y Scribner, K. T. (2019). 18S rRNA metabarcoding diet analysis of a predatory fish community across seasonal changes in prey availability. Ecology and Evolution, 9, 1410–1430. https://doi.org/10.1002/ece3.4857
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R. y Hebert, P. D. N. (2005). DNA barcoding Australia’s fish species. Philosophical Transactions of the Royal Society B: Biological Sciences, 360, 1847–1857. https://doi.org/10.1098/rstb.2005.
1716
White, T. J., Bruns, T., Lee, S. y Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. En M. A. Innis, D. H. Gelfrand, J. J. Snisky y J. J. White (Eds.), PCR protocols: a guide to methods and applications (pp. 315–320). San Diego: Academic Press.
Yoo, T. H., Kang, H. E., Lee, S. R., Lee, J. B., Baeck, G. W., Park, H. et al. (2017). Metabarcoding analysis of the stomach contents of the Antarctic Toothfish (Dissostichus mawsoni) collected in the Antarctic Ocean. PeerJ, 5, e3977. https://doi.org/10.7717/peerj.3977
Yu, J., Xue, J. H. y Zhou, S. L. (2011). New universal matK primers for DNA barcoding angiosperms. Journal of Systematics and Evolution, 49, 176–181. https://doi.org/10.1111/j.1759-6831.2011.00134.x


